






微生物生物地理学研究方法
Research Methods of Microbial Biogeography
彭子恒1,焦硕1, *,韦革宏1
1西北农林科技大学生命科学学院,旱区作物逆境生物学国家重要实验室,陕西省农业与环境微生物重点实验室,陕西杨凌
*通讯作者邮箱:shuojiao@nwafu.edu.cn
彭子恒, 焦硕, 韦革宏. (2021). 微生物生物地理学研究方法. 微生物组实验手册. Bio-101: e2003930. DOI: 10.21769/BioProtoc.2003930.
How to cite: Yunyun Gao, Kai Peng, Defeng Bai, et al. 2024. The Microbiome Protocols eBook initiative: Building a bridge to microbiome research. iMeta 3: e182. https://doi.org/10.1002/imt2.182
摘要:微生物生物地理研究对于理解微生物多样性的形成和维持机制至关重要。目前高通量测序的突破为我们探究微生物地理分布格局提供了支持。本文以河西走廊森林土壤细菌的地理分布研究为示例,从纬度多样性模式、alpha-多样性的影响因子、群落结构的影响因子、距离衰减关系和群落生态过程等研究方法入手,阐述河西走廊森林土壤细菌的生物地理分布模式和群落构建机制,给出了微生物生物地理分布的常见研究方法和分析流程,为研究微生物生物地理学提供了科学案列和参考。
关键词:生物地理学;细菌群落;16S rRNA;空间分布;河西走廊
背景介绍:生物地理学是一门研究生物多样性在空间和时间上分布格局的生物学与地理学交叉学科,主要探究生物群落的组成、分布和形成原因。生物地理学的研究为生物多样性的形成和维持机制提供了见识,例如物种形成,灭绝,扩散和物种相互作用,并且有助于预测生态系统功能随环境的变化。生物地理学按照生物群落可以划分为植物生物地理、动物生物地理和微生物生物地理。当前,随着高通量技术的发展,微生物生物地理学迎来了发展机遇。
仪器设备
1.普通个人电脑
软件
1.R (v3.6.2),所需依赖包:ggplot2、reshape2、patchwork、vegan、geosphere、psych、corrplot和pacman。
实验步骤
一、数据准备
本分析以河西走廊数据森林土壤细菌群落数据为示例,共28个样本,包含6个数据表,分别是细菌群落数据(OTU绝对丰度表)、细菌物种分类数据、土壤理化数据、气候数据、样点的经纬度数据、βNTI矩阵数据。
1.数据导入
env <- read.csv("env.csv", row.names = 1) #土壤理化数
geo <- read.csv("geo.csv", row.names = 1) #样点的经纬度数据
otu <- read.csv("otu.csv", row.names = 1) #细菌群落数据
taxon <- read.csv("taxon.csv", row.names = 1) #细菌物种分类数据
clim <- read.csv("clim.csv", row.names = 1) #气候数据
bnti <- read.csv("nti.csv", row.names = 1) # βNTI数据
2.加载包
library(pacman)
p_load(ggplot2,reshape2,patchwork,vegan,geosphere,psych,corrplot)
二、细菌物种组成
1.分析介绍:物种组成分析是最基本的生物地理模式之一。它主要回答了物种在何处分布以及如何分布的问题。
2.分析目的:探究河西走廊数据森林土壤细菌群落在主要门水平的分布
3.分析方法:堆叠柱形图
4.数据分析
otu_abun <- otu/colSums(otu) #转换为相对丰度
p<- aggregate(otu_abun, by=list(taxon$Phylum), sum) #门水平
rownames(p) <- p$Group.1
p <- p[,-1]
top <- names(head(sort(rowSums(p), decreasing = T), 57)) #基于相对丰度进行排序
top_10<- c("Proteobacteria", "Actinobacteria", "Acidobacteria", "Chloroflexi", "Gemmatimonadetes","Bacteroidetes", "Planctomycetes","Firmicutes", "Thermomicrobia", "Nitrospirae")
taxon$Phylum <- as.character(taxon$Phylum)
taxon$Phylum[!(taxon$Phylum)%in%top_10] <- "Others"
p_top <- aggregate(otu_abun, by=list(taxon$Phylum), sum) #获取top10
rownames(p_top) <- p_top[,1]
p_top <- p_top[,-1]
p_top <- p_top[order(rowSums(p_top)),] #按每行和排序
q_top <- t(p_top)
q_top <- as.data.frame(q_top)
q_top$sample <- rownames(q_top)
q <- melt(q_top,ID="names")
colnames(q)[names(q)=="variable"]<-"Taxa"
q$factor <- "forest"
5.数据绘图
colors<-c("grey50","darkolivegreen3","gold","dodgerblue4","darkseagreen",< span="">
"chartreuse4","darkorange","burlywood2","brown3","#984EA3","cyan3")
ggplot(q, aes( x = sample, y = value, fill = Taxa))+
geom_bar(position = "fill", stat = "identity")+
theme_bw()+
scale_fill_manual(values=colors)+
scale_y_continuous(expand = c(0,0))+
labs(x="",y="Relative Abundance",fill="Phylum")+
theme(text=element_text(size=12),
axis.text.y=element_text(size=12,color = "black"),
axis.text.x=element_text(size=12,color = "black",angle = 45, hjust = 0.5, vjust = 0.5),
legend.title=element_text(size=12),
legend.text=element_text(size=12))+
theme(panel.grid = element_blank(),
panel.background = element_rect(color = 'black', fill = 'transparent')) +
guides(fill=guide_legend(keywidth = 1.3, keyheight = 2))
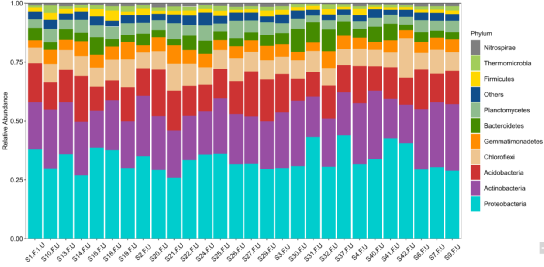 |
图1. 细菌群落物种丰度堆叠柱形图
6.分析结果
变形菌门和放线菌门是丰度最高的两个门。
三、纬度多样性模式
1.分析介绍:纬度多样性模式是最重要的生物地理模式之一,即随着纬度的增加,物种多样性逐渐减小。目前微生物多样性的纬度梯度模式已经得到了广泛的研究。
2.分析目的:探究河西走廊数据森林土壤细菌群落随纬度的多样性模式
3.分析方法:线性拟合图
4.数据分析
otu <- as.data.frame(t(otu))
richness <- specnumber(otu) #计算丰富度
shannon <- diversity(otu,index = "shannon") #计算shannon多样性
diversity <- data.frame(richness,shannon)
aa <- cbind(geo, diversity)
summary(lm(aa$richness ~ aa$lat))
summary(lm(aa$shannon ~ aa$lat))
表1. 纬度与细菌alpha-多样性的线性回归
Richness | Shannon | |||||
Slope | R2 | P | Slope | R2 | P | |
Latitude | -103.64 | 0.105 | 0.051 | -0.106 | 0.232 | < 0.01 |
5.数据绘图
p1 <- ggplot(aa,aes(x = lat, y = richness))+
geom_point()+
geom_smooth(method = "lm",alpha = 0.2)
p2 <- ggplot(aa,aes(x = lat, y = shannon))+
geom_point()+
geom_smooth(method = "lm",alpha = 0.2)
plot <- p1 + p2
plot
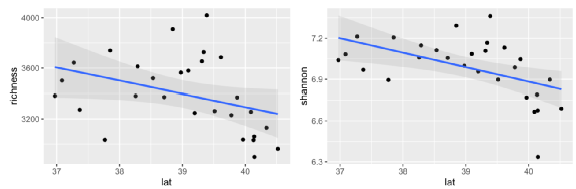 |
图2. 细菌α-多样性随纬度的变化
6.分析结果
细菌群落多样性随着纬度的增加而下降,说明存在纬度多样性模式。
四、细菌α-多样性的影响因素
1.分析介绍:探究微生物多样性的影响因子主要是回答多样性为什么这样分布。微生物多样性的影响因子主要从土壤因子和气候因子两方面考虑。
2.分析目的:探究影响细菌多样性的主要环境因子
3.分析方法:相关性分析,通过热图展示
4.数据分析
diversity <- data.frame(richness,shannon)
cor_soil <- corr.test(diversity, env[,1:14], method = "spearman", adjust = "none")
cor_soil_r <- cor_soil$r
cor_soil_p <- cor_soil$p
cor_soil_p[cor_soil_p >= 0.05] <- -1
cor_soil_p[cor_soil_p < 0.05 & cor_soil_p >= 0] <- 1
cor_soil_p[cor_soil_p == -1] <- 0
corr_soil <- cor_soil_r*cor_soil_p
cor_clim <- corr.test(diversity, clim[,1:19],method = "spearman",adjust = "none")
cor_clim_r <- cor_clim$r
cor_clim_p <- cor_clim$p
cor_clim_p[cor_clim_p >= 0.05] <- -1
cor_clim_p[cor_clim_p < 0.05 & cor_clim_p >= 0] <- 1
cor_clim_p[cor_clim_p == -1] <- 0
corr_clim <- cor_clim_r*cor_clim_p
5.数据绘图
col<-colorRampPalette(c("#77AADD","#4477AA","#FFFFFF", "#EE9988","#BB4444"))
corrplot(corr_soil, method = 'color', addCoef.col = 'black', number.cex = 0.8, rect.col = "black",addgrid.col = "black",col = col(200),tl.col = 'black', cl.pos = "n")
corrplot(corr_clim, method = 'color', addCoef.col = 'black', number.cex = 0.8, rect.col = "black",addgrid.col = "black",col = col(200),tl.col = 'black', cl.pos = "n")
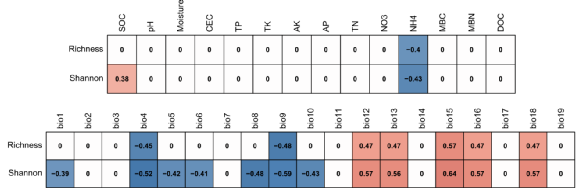 |
图3. 细菌多样性与环境因子的相关性
6.分析结果
细菌多样性与NH4呈显著负相关,与bio4 (Temperature seasonality) 显著负相关,与bio12 (MAP) 显著正相关。
五、距离衰减关系
1.分析介绍:群落组成相似性的距离衰减是生物地理学的基本模式之一,即群落相似性随着地理距离的增加而下降。
2.分析目的:探究河西走廊森林土壤细菌群落的距离衰减关系,即细菌群落的相似性是否随着地理距离的增大而降低
3.分析方法:线性拟合图
4.数据分析
d.geo <- distm(geo, fun = distHaversine) #经纬度转换为地理距离
dist.geo <- as.dist(d.geo)
dist_geo <- as.data.frame(as.vector(dist.geo))
dist_geo <- dist_geo/1000 #地理距离单位转换为千米
colnames(dist_geo) <- "dist_geo"
dist.otu <- vegdist(otu, method = "bray")
dist_otu <- as.data.frame(as.vector(dist.otu))
colnames(dist_otu) <- "dist_otu"
data <- data.frame(dist_geo, dist_otu)
data$dist_otu <- 1-data$dist_otu
data$dist_otu <- data$dist_otu * 100 #转换为百分比
summary(lm(data$dist_otu ~ data$dist_geo))
表2. 细菌距离衰减关系的线性回归
Slope | R2 | P | |
Distance decay | -0.015 | 0.203 | < 0.001 |
5.数据绘图
ggplot(data, aes(x = dist_geo,y = dist_otu)) +
geom_point() +
geom_smooth(method = "lm",alpha = 0.2) +
labs(x = "Geographic Distance (Km)", y = "Community Similarity (%) ")
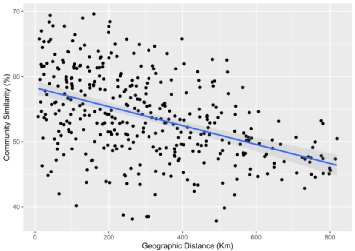 |
图4. 距离衰减关系
6.分析结果
河西走廊森林土壤细菌群落呈现出显著的距离衰减关系。
六、细菌群落结构的影响因素
1.分析介绍:探究微生物群落结构的影响因子主要是回答细菌群落为什么这样分布。微生物群落结构的影响因子主要从土壤因子和气候因子两方面考虑。
2.分析目的:探究影响细菌群落结构的主要环境因子
3.分析方法:曼特尔检验 (Mantel test)
4.数据分析
dist.otu <- vegdist(otu, method = "bray")
soil <- NULL
for (i in 1:ncol(env)) {
aa <- mantel(dist.otu, dist(env[,i], method = "euclidean"), method = "pearson", permutations = 9999, na.rm = TRUE)
soil <- rbind(soil,c(colnames(env)[i],aa$statistic, aa$signif))
}
climate <- NULL
for (i in 1:ncol(clim)) {
aa <- mantel(dist.otu, dist(clim[,i],method = "euclidean"), method = "pearson", permutations = 9999, na.rm = TRUE)
climate <- rbind(climate,c(colnames(clim)[i],aa$statistic, aa$signif))
}
5.数据展示
表3. 细菌群落与环境因子的曼特尔检验
Mantel test | |||||
Soil | R | P | Climate | R | P |
SOC | 0.090 | 0.236 | Bio1 | 0.094 | 0.199 |
pH | 0.013 | 0.424 | Bio2 | 0.156 | 0.120 |
Moisture | 0.098 | 0.179 | Bio3 | 0.012 | 0.419 |
CEC | 0.243 | 0.008 | Bio4 | 0.246 | 0.032 |
TP | -0.043 | 0.638 | Bio5 | 0.177 | 0.083 |
TK | 0.012 | 0.419 | Bio6 | 0.010 | 0.382 |
AK | 0.127 | 0.140 | Bio7 | 0.186 | 0.100 |
AP | -0.127 | 0.838 | Bio8 | 0.146 | 0.122 |
TN | 0.121 | 0.145 | Bio9 | 0.158 | 0.086 |
NO3 | 0.228 | 0.058 | Bio10 | 0.132 | 0.138 |
NH4 | 0.007 | 0.437 | Bio11 | 0.033 | 0.357 |
MBC | -0.093 | 0.755 | Bio12 | 0.172 | 0.112 |
MBN | 0.016 | 0.400 | Bio13 | 0.221 | 0.059 |
DOC | -0.001 | 0.433 | Bio14 | 0.060 | 0.277 |
Bio15 | 0.102 | 0.220 | |||
Bio16 | 0.193 | 0.082 | |||
Bio17 | 0.086 | 0.256 | |||
Bio18 | 0.203 | 0.077 | |||
Bio19 | 0.074 | 0.261 |
6.分析结果
细菌群落结构与CEC (Cation Exchangeable Capacity)和bio4 (Temperature seasonality) 呈显著相关性。
七、细菌群落结构β多样性
1.分析介绍:探究微生物群落结构β多样性分布模式是生物地理模式的基本问题。
2.分析目的:探究细菌群落结构随CEC的分布模式
3.分析方法:PCoA 主坐标分析 (principal coordinate analysis)
4.数据分析
pcoa <- cmdscale(dist.otu, k = 3, eig = TRUE)
pcoa_eig <- (pcoa$eig)[1:2] / sum(pcoa$eig)
site <- data.frame(pcoa$points)[1:2]
site <- cbind(site,env$CEC)
colnames(site) <- c('PCoA1', 'PCoA2',"CEC")
5.数据展示
ggplot(site, aes(PCoA1, PCoA2)) +
geom_point(aes(color = CEC),size = 3)+
scale_color_gradient(low = "blue",high = "red") +
labs(x = paste('PCoA1: ', round(100 * pcoa_eig[1], 2), '%'),
y = paste('PCoA2: ', round(100 * pcoa_eig[2], 2), '%'))
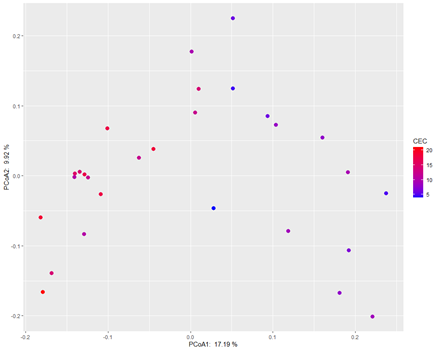
6.分析结果
细菌群落β多样性随土壤CEC呈现出明显的变化。
八、群落生态过程
1.分析介绍:生态群落构建过程通常分为基于生态位的确定性过程和基于中性模型的随机性过程。目前扩散限制和环境选择已被广泛接受为驱动微生物分布的两个主要因素。
2.分析目的:探究空间和环境因子对河西走廊森林土壤细菌群落分布的影响
3.分析方法:null model by (Stegen, 2013);饼图
## Beta_NTI计算方法
library(picante)
commu <- read.csv("otu.csv",fileEncoding = "UCS-2LE",row.names = 1)
phylo <- read.tree("OTUs.tre") #读入系统发育树
Beta_NTI<-function(phylo,comun,beta.reps=999){< span="">
comun=t(comun)
match.phylo.comun = match.phylo.data(phylo, t(comun))
beta.mntd.weighted = as.matrix(comdistnt(t(match.phylo.comun$data),cophenetic(match.phylo.comun$phy),abundance.weighted=T))
rand.weighted.bMNTD.comp = array(c(-999), dim=c(ncol(match.phylo.comun$data), ncol(match.phylo.comun$data),beta.reps))
for (rep in 1:beta.reps) {
rand.weighted.bMNTD.comp[,,rep] = as.matrix(comdistnt(t(match.phylo.comun$data),taxaShuffle(cophenetic(match.phylo.comun$phy)),abundance.weighted=T,exclude.conspecifics = F))
print(c(date(),rep))
}
weighted.bNTI = matrix(c(NA),nrow=ncol(match.phylo.comun$data),ncol=ncol(match.phylo.comun$data))
for(columns in 1:(ncol(match.phylo.comun$data)-1)) {
for(rows in (columns+1):ncol(match.phylo.comun$data)) {
rand.vals = rand.weighted.bMNTD.comp[rows,columns,];
weighted.bNTI[rows,columns] = (beta.mntd.weighted[rows,columns] - mean(rand.vals)) / sd(rand.vals)
rm("rand.vals")
}
}
rownames(weighted.bNTI) = colnames(match.phylo.comun$data);
colnames(weighted.bNTI) = colnames(match.phylo.comun$data);
return(as.dist(weighted.bNTI))
}
bnti <- Beta_NTI(phylo,comun,beta.reps=999)
注:Beta_NTI计算需要在服务器中进行,这里只给出计算方法。
4.数据分析
bnti <- bnti[lower.tri(bnti)]
sto <- length(which(abs(bnti)<2))< span="">
det <->2))
assem <- c(sto,det)
5.数据展示
pie(assem, labels=c("stochastic process","deterministic process"),col = c("#F8766D","#00BFC4"),radius = 1)
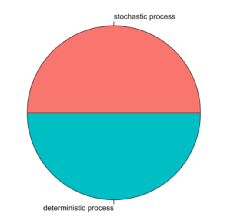
图5. 生态群落构建过程
6.分析结果
确定性过程 (50%) 和随机性过程 (50%) 共同解释了细菌群落的分布。
致谢
本项工作得到了国家自然科学基金委员会 (项目编号:41830755和41807030) 的资助。
参考文献
1.Stegen, J. C., Lin, X., Fredrickson, J. K., Chen, X., Kennedy, D. W., Murray, C. J., Rockhold, M. L. and Konopka, A. E. 2013. Quantifying community assembly processes and identifying features that impose them. The ISME Journal 7(11): 2069-2079.
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 2423
2423

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









