






肺炎患者痰液微生物组成与疾病严重程度相关
Highly diverse sputum microbiota correlates with the disease severity in patients with community-acquired pneumonia: a longitudinal cohort study

Article,2024-05-29,Respiratory Research,[IF 4.7]
DOI:10.1186/s12931-024-02821-2
原文链接:https://respiratory-research.biomedcentral.com/articles/10.1186/s12931-024-02821-2
第一作者:Jing Yang (杨景), Jinman Li (李金蔓), Linfeng Zhang (张林峰)
通讯作者:Lili Ren (任丽丽), Mingkun Li (李明锟), Jianwei Wang (王健伟)
主要单位:
国家生物信息中心
中国医学科学院病原生物学研究所
- 背景 -
人体呼吸道微生物组在维持肺部生态和免疫稳态中起关键作用,影响肺部健康和对病原感染的易感性。在新冠大流行之前,包括社区获得性肺炎(Community-acquired pneumonia, CAP)在内的下呼吸道感染是全球造成人口死亡的第四大病因。尽管病原感染是CAP的主要原因,但只有不到50%的患者中能够检测到特定致病微生物。已有研究发现,CAP患者的呼吸道微生物组比健康人群多样性低,且富含致病菌,微生物可能通过调节免疫系统影响肺炎严重程度。然而,现有研究主要集中在高风险群体,样本量小且多为横断面研究,未能全面揭示免疫正常成年人中的情况。
- 导读 -
近期,国家生物信息中心李明锟团队、中国医学科学院病原生物学研究所任丽丽团队、深圳市第三人民医院、哈尔滨医科大学附属第二医院、西安交通大学第一附属医院、东南大学附属中大医院团队合作在《Respiratory Research》发表了研究论文“Highly diverse sputum microbiota correlates with the disease severity in patients with community-acquired pneumonia: a longitudinal cohort study”,该研究通过分析来自中国6个不同省份医院350个CAP患者的917个痰液样本16S rRNA测序数据,探讨了痰液微生物组与疾病严重程度的关系,为CAP的发病机制和诊疗提供了新思路。研究发现71% 样本的痰液微生物组主要由呼吸道共生菌组成,15%的样本以五种机会致病菌为主,而5%的样本与阴性对照组的组成相似(提示低菌量)。与非重症CAP患者相比,重症患者的痰液微生物组更多被潜在的病原菌主导、与健康状态的偏差更大、住院期间的变化更显著、细菌间的相互作用网络更稀疏。研究发现入院时的痰液微生物组对疾病严重程度有一定的预测作用(AUC=0.74)。此外,还发现不同的病原菌感染与特定的微生物组改变有关。
国家生物信息中心博士生杨景和硕士毕业生张林峰,中国医学科学院病原生物所博士毕业生李金蔓为本文的共同第一作者,国家生物信息中心李明锟研究员和中国医学科学院病原生物所任丽丽研究员、王健伟研究员为本文的共同通讯作者。
本研究拟回答的科学问题
肺炎病人呼吸道微生物组是否与健康人存在差异?
肺炎病人呼吸道微生物组是否由机会致病菌主导?
肺炎病人呼吸道微生物组是否与疾病严重程度相关?
不同病原导致的肺炎病人呼吸道微生物组是否相同?
- 主要内容 -
1. 样本和测序数据概览
本研究纳入纵向痰样1,065 份,来自中国不同省市六家医院的367名确诊CAP的住院患者。经过测序数据的质量控制后,来自350名患者的917份样本和25份阴性对照(NCs)样本被用于后续分析。CAP患者的痰液微生物组组成与NCs显著不同,NCs中的主要组分在CAP患者中占比很少,表明背景污染极少。同时,本研究使用了来自三项先前研究的876名中国健康个体的痰液微生物组作为健康对照组(HCs)。
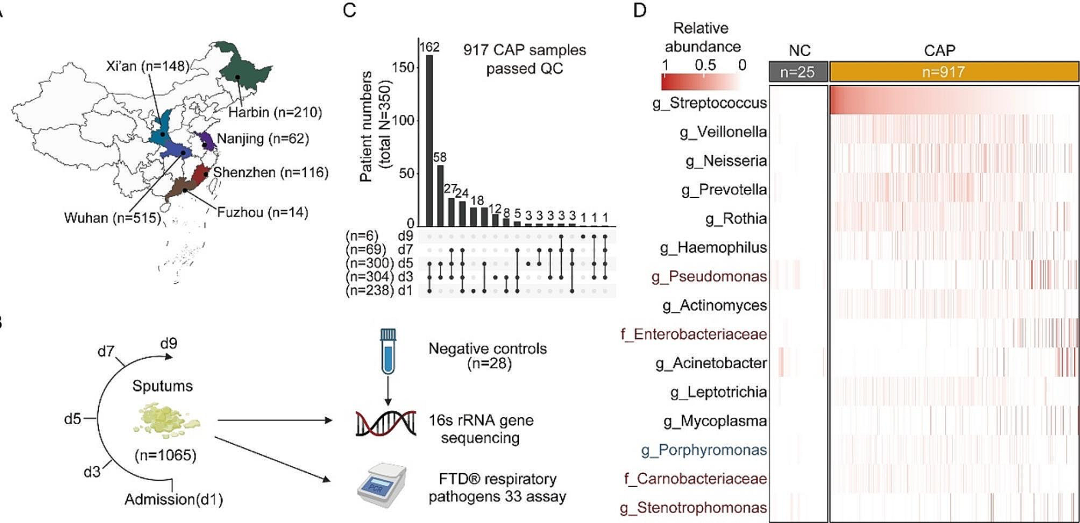
图1. 队列设计和痰液微生物组成
2. CAP患者痰液微生物组成
在CAP患者的痰液中,六种常见的呼吸道共生微生物(链球菌、韦荣菌、奈瑟菌、普雷沃氏菌、罗氏菌、嗜血杆菌)丰度最高,占CAP微生物序列总数的51.2%、健康对照微生物序列总数的38.0%,以及阴性对照序列总数的1.4%。CAP患者的痰液微生物组多样性和组成显著不同于HCs,可能的致病菌(如假单胞菌,肠杆菌科,鞘氨醇单胞菌,窄食单胞菌)在CAP样本中显著富集。不同个体间的痰液微生物组表现出显著异质性,聚类分析发现了九个不同的聚类(CSs),其中CS2-4(占CAP患者的71.1% )以多种共生细菌为主,α多样性较高,且与健康对照菌群组成更相似;CS1、CS5和CS7-9则以单一的机会致病菌为主,α多样性较低,且与健康对照差异更大;CS6痰液样本微生物载量相对较低,倾向于出现在特定患者中,与住院后治疗无关。CS2-4患者的重症率为12.9%,与CS6相似,但显著低于CS1、5、7、8、9(36.4%)。
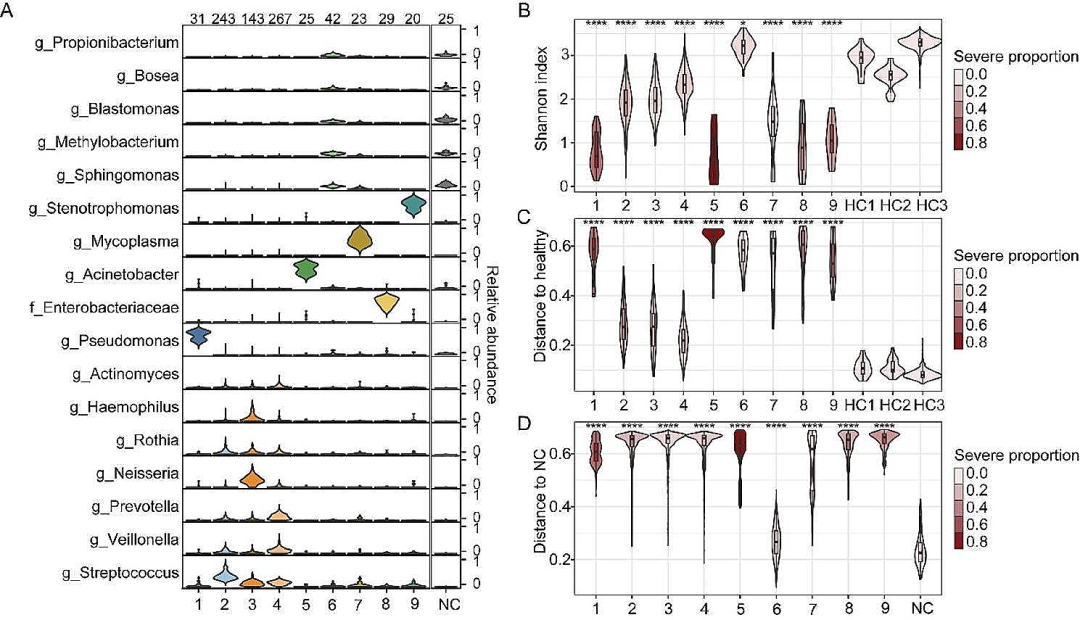
图2. CAP患者痰液微生物组成的分型
3. 痰液微生物组与疾病严重程度的关联
为减少入院后抗生素和其他医疗干预对痰液微生物组成的影响,我们仅分析了入院第一天收集的238个样本。结果显示,疾病严重程度指标(CURB65评分、PSI评分、吸氧时长、住院时间、临床结局和严重程度诊断)与痰液微生物组成显著相关。重症患者的痰液微生物组α多样性较低,53.8%的重症患者微生物组成由一种潜在的致病菌主导(丰度超过50%)。肠杆菌科在重症患者中富集,且与吸氧时长正相关。基于痰液微生物组成数据训练的分类器能较好区分重症和非重症病例(AUC=0.74)。肠杆菌科贡献的甲萘醌生物合成通路在重症病例中富集,而卟啉单胞菌和梭杆菌贡献的丁酸盐发酵通路在非重症病例中富集。
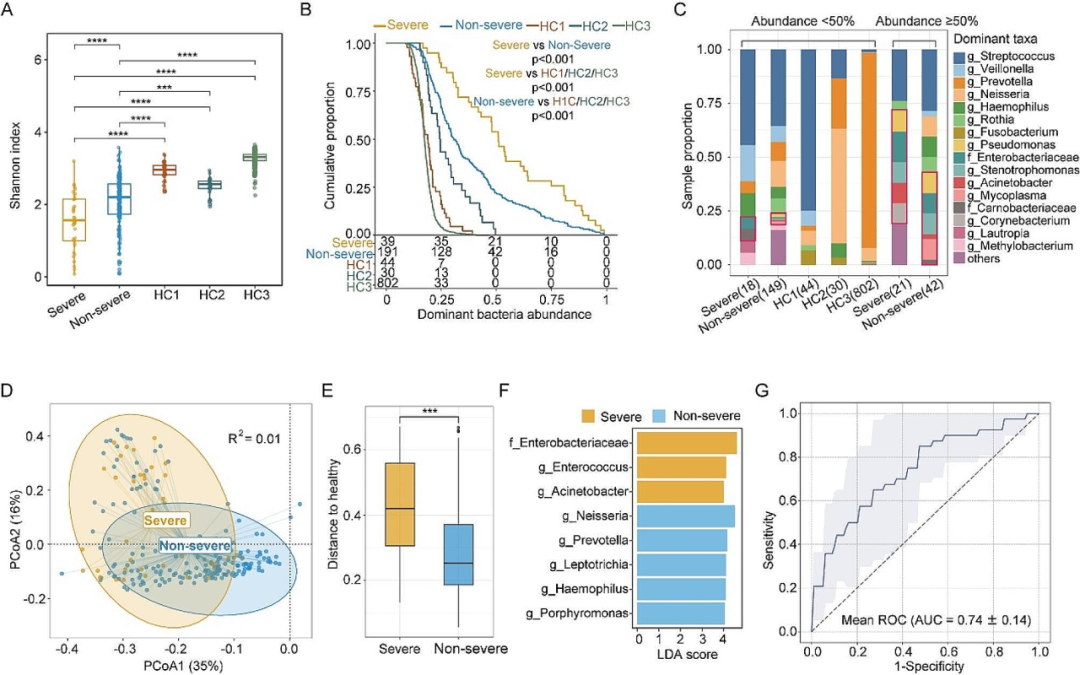
图3. 不同严重程度的 CAP 患者与健康人之间痰液微生物组差异
4. 痰液微生物组的动态变化及其与疾病严重程度的关联
我们研究了非重症和重症病例中痰液微生物组在住院期间变化的差异。结果显示,重症病例痰液微生物组成在连续时间点间变化较大、与初始状态组成差异更大。重症病例中CS转换更频繁,尤其是第1天CS8样本在第5天全部转换为CS5,这种转换可能与侵入性机械通气后的二次感染有关。重症患者痰液中细菌间相互作用网络更为稀疏,且在住院期间有向更稀疏状态发展的趋势,网络中包含更多潜在致病菌,表明其组成相较非重症患者更加紊乱。住院期间,无论重症还是非重症患者的痰液微生物组都未向健康状态转换。
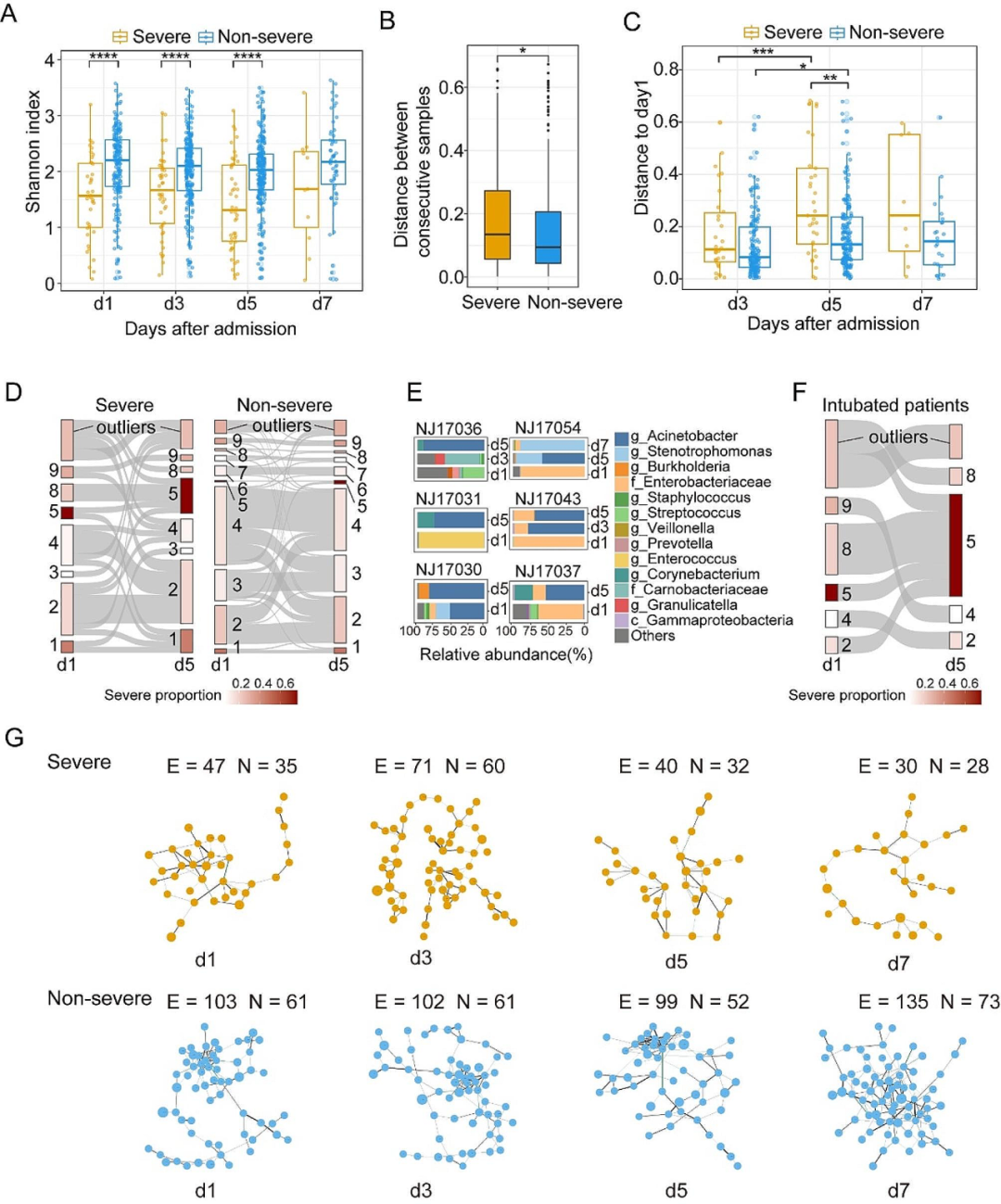
图4. 痰液微生物组的动态及其与疾病严重程度的关系
5. 不同病原体感染患者的痰液微生物组差异
使用FTD®呼吸道病原检测试剂盒在256名患者的548个样本中鉴定出可能的病原体。为避免二次感染的影响,随后分析仅限于入院后三天内FTD结果阳性的216名患者。90名患者疑似感染细菌,88名患者疑似感染病毒,38名患者为细菌、病毒混合感染。我们发现细菌感染与病毒感染、病毒感染与混合感染之间的微生物组组成存在显著差异,且这三种情况的微生物组均与健康对照组显著不同,其中细菌感染样本的偏离度更大。根据病原微生物将感染病人分组,仅考虑感染人数超过15的高发病原(鼻病毒、肺炎支原体、肺炎克雷伯菌和甲型流感),并排除混合感染样本。首先,我们发现试剂盒检出的病原不一定是患者痰液微生物组的主导物种,如肺炎支原体感染患者中,只有3名患者的痰液微生物组是以肺炎支原体为主,其余主要由呼吸道共生菌主导。尽管不同病原感染患者痰液微生物组的α多样性无显著差异,但肺炎支原体感染的微生物组组成与肺炎克雷伯菌感染、鼻病毒感染显著不同,且在不同病原类型样本中富集了不同的细菌,例如,鼻病毒感染样本中肠球菌和窄食单胞菌相对丰度较高,甲型流感感染样本中不动杆菌和假单胞菌丰度较高,肺炎支原体感染样本中罗氏菌和肉杆菌科丰度较高,肺炎克雷伯菌感染样本中不动杆菌丰度较高。
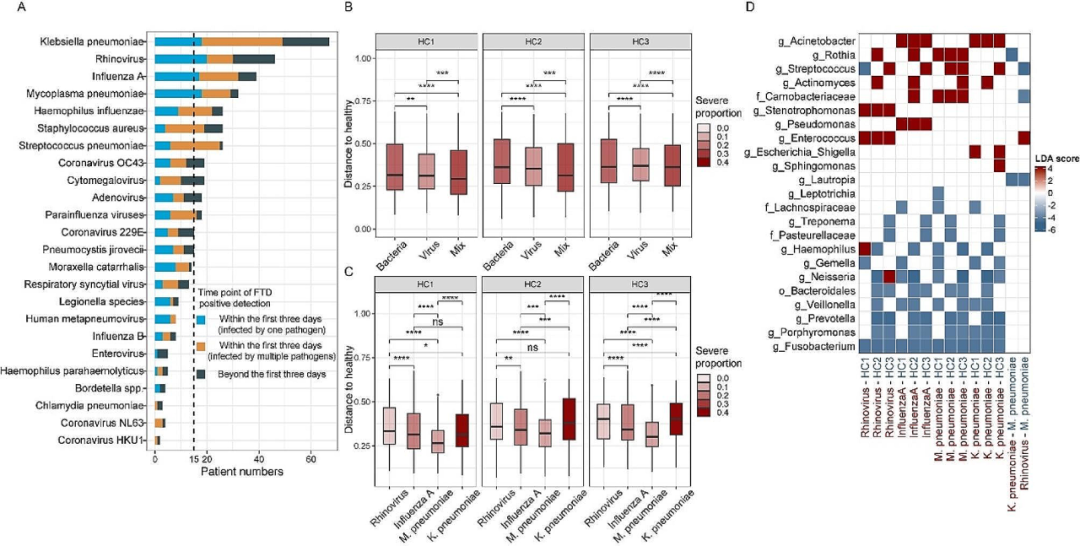
图5. 感染不同病原体患者的微生物组特征
- 总结 -
我们的研究表明,社区获得性肺炎(CAP)患者的痰液微生物组组成多样,其中大部分患者,尤其是非重症患者的痰液微生物组与健康个体相似。重症CAP病例痰液微生物组更多由潜在致病菌主导,并在住院期间经历更大的变化。未来需要进一步开展前瞻性和干预性研究,揭示呼吸道微生物组变化与疾病严重程度之间的因果关系。
参考文献
Yang, J., Li, J., Zhang, L. et al. Highly diverse sputum microbiota correlates with the disease severity in patients with community-acquired pneumonia: a longitudinal cohort study. Respir Res25, 223 (2024). https://doi.org/10.1186/s12931-024-02821-2
- 通讯作者简介 -

中国医学科学院
病原生物学研究所
任丽丽
研究员
任丽丽,研究员,博士生导师。中国医学科学院呼吸道疾病病原组研究重点实验室主任。长期致力于呼吸道感染病原组和致病机制研究,入选国家高层次人才支持计划。近五年以第一和通讯(含共同)作者在Lancet、JAMA、Cell Host microbe和Am J Respir Crit Care Med等期刊发表SCI论文多篇。连续入选科睿唯安2022年度和2023年度全球高被引科学家。

中国科学院
国家生物信息中心
李明锟
研究员、博士生导师
研究员、博士生导师、应用发展部部长,长期致力于高通量测序数据深度挖掘与基因组变异及进化分析,入选国家海外高层次人才青年项目,北京重大呼吸道传染病研究中心专家委员会委员,中华预防医学会生物信息学分会和中国医疗保健国际交流促进会临床微生物学分会常务委员。研究方向包括:临床宏基因组技术研发,人体微生物组特征及其与营养、疾病关联,病原微生物基因组变异与演化分析。近五年以通讯(含共同)作者在Nature Ecology Evolution、Cell Host microbe、Am J Respir Crit Care Med等期刊发表SCI论文25篇,被引用超过2900次。

中国医学科学院
王健伟
教授(长聘)、研究员
王健伟,研究员,博士生导师。副院校长。国家基金委杰青、创新群体负责人,长江学者,中国医学科学院学部委员。中法新发病原体联合实验室主任。长期致力于呼吸道感染致病机制和应用策略研究,近五年以通讯(含共同)作者在Lancet、Cell、Cell Host microbe和Am J Respir Crit Care Med等期刊发表SCI论文多篇。连续入选科唯睿安全球高被引科学家和爱斯唯尔高被引学者。
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文,跳转最新文章目录阅读


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









