






点击蓝字 关注我们
培养和非培养方法揭示特定细菌在人类皮肤老化中的作用

研究论文
● 原文链接DOI: https://doi.org/10.1002/imo2.26
●2024年9月11日,清华大学刘晓团队和复旦大学王久存团队在iMetaOmics在线发表了题为“Culture-dependent and -independent approaches reveal the role of specific bacteria in human skin aging”的文章。
● 本研究对来自中国人群的 822 份面部微生物样本及其相应部位的 14 种皮肤表型进行了关联分析,鉴定出多种与表型相关的微生物物种。随后从皮肤中分离出三种关键物种并使用其培养滤液与皮肤角质形成细胞、成纤维细胞互作,结果显示奥斯陆莫拉菌能调节胶原代谢、细胞外基质的组装、并参与多个细胞衰老过程。本项研究可为皮肤抗衰老干预提供新思路。
● 第一作者:夏晶晶、钟倩、李志明、魏情珍
● 通讯作者:刘晓(liuxiao@sz.tsinghua.edu.cn)、王久存(jcwang@fudan.edu.cn)、吉恩.克鲁德曼(Jean.Krutmann@IUF-Duesseldorf.de)
● 合作作者:蒋刘一琦、段程、贾慧珏、谈益妹、韩涟漪
● 主要单位:复旦大学生命科学学院、清华大学深圳国际研究生院、粤港澳大湾区精准医学研究院(广州)
亮 点

● 利用鸟枪法宏基因组测序数据集分析了与年龄和衰老表型相关的皮肤微生物组动态;
● 通过机器学习建模,预测奥斯陆莫拉菌Moraxella osloensis和痤疮丙酸杆菌Cutibacterium acnes是早衰/延迟衰老相关的微生物物种;
● 从健康皮肤中分离出的奥斯陆莫拉菌Moraxella osloensis在体外调节胶原蛋白代谢和细胞外基质组装;
● 与细菌群落相比,年龄对真菌群落变化的影响相对较小。本研究分析了与年龄相关的真菌类群。
摘 要
皮肤老化是一个动态过程,涉及一系列表型变化,因此是研究微生物组-表型相互作用的一个有吸引力的模型。因此,我们对中国人群中的 822 份面部微生物样本和相应部位的 14 种皮肤表型进行了评估。卟啉和年龄显示了最显著的微生物变异。我们进一步分析了与年龄和衰老表型相关的皮肤微生物组的动态变化。通过多元线性回归模型,我们预测了与早衰/延迟衰老相关的微生物种类,主要是奥斯陆莫拉菌和痤疮丙酸杆菌。我们还在体外验证了宿主-微生物相互作用的生物学功能。从健康皮肤中分离出的奥斯陆莫拉菌能调节胶原代谢和细胞外基质的组装,并能促进人类角质形成细胞和成纤维细胞的衰老,因此有可能用于抗衰老干预措施的开发。
视频解读
Bilibili:https://www.bilibili.com/video/BV1eW44exEgm/
Youtube:https://youtu.be/0DrtfbAqeis
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
皮肤上有数百万种不同的微生物,包括细菌、真菌和病毒,统称为人类皮肤微生物组。这些共生菌对维持皮肤平衡非常重要,皮肤微生物组的破坏与一些最常见皮肤病的各种临床症状有关。
即使是健康人,皮肤微生物群也是高度个性化和动态的。影响皮肤细菌群落的变量很多,包括宿主因素(如种族、性别和年龄)、职业或生活方式(如饮食、卫生、护肤品和药物)或其他暴露(如气候、地理位置、污染、紫外线和其他辐射)造成的环境因素。不过,从传统生态学的角度来看,由于皮肤被视为一个生态系统,塑造生态系统的选择压力可简化为三个主要因素: 1)资源可用性(营养物质的存在);2)环境条件(温度和地理位置);3)生物因素(通过自我适应或微生物与微生物之间的相互作用占据生态位)。以往的证据也有力地表明,不同生态位的物理和化学景观主要驱动着当地微生物的组成(生态位选择)。反过来,这些微生物的生长和新陈代谢又会改变当地的生态位,如 pH 值或湿度状态,并重塑宿主皮肤的表型。然而,对皮肤微生物组-表型相互作用的理解仍然模糊不清,需要系统的特征描述。
皮肤老化是一个动态过程,在外观和功能方面会出现一系列与年龄相关的表型变化,因此是研究微生物组-表型相互作用的理想模型。尽管许多研究已经证实了时序年龄对皮肤细菌群落的影响,但对皮肤衰老引起的跨界微生物群变异的了解仍然很少。然而,这并不是一件小事。虽然在大多数情况下,年龄与皮肤衰老表型有很好的相关性,反之亦然,但由于皮肤衰老是一个结合了时间衰老和外在衰老的复杂过程,人体“肤龄”可能与时序年龄有很大偏差。
为了探索跨界微生物组与皮肤表型的相互作用,研究人员从上海招募了294名健康人,这是迄今为止在中国人群中进行皮肤宏基因组分析的最大样本量。研究人员收集了前额(FH)、面颊(CK)和鼻旁(NS)三个面部部位的皮肤微生物组。利用鸟枪法宏基因组测序技术对总共 822 个样本进行了评估,从而对所有微生物群(细菌、真菌和病毒)进行了更精确的调查。研究人员交叉分析了多种潜在的宿主变量,包括年龄、性别和 12 种皮肤表型,以阐明健康中国个体中宿主与微生物群之间的相互作用。与以往的研究相比,本研究更进一步;除了探讨衰老与微生物群的相关性外,我们还使用二维(2D)培养体系来验证了这些关联。研究的一般工作流程如图 1A 所示。
结 果
时序年龄在皮肤表型中起关键作用,并解释了皮肤细菌的巨大差异
对相应采样区域的皮肤微生物组和表型(皮肤特征集合)进行了评估(图 1A)。由于解剖结构的限制,没有收集鼻旁部位的一些参数。不出所料,年龄对皮肤外观和生理有重要影响(图 1A,图 S1A-C)。随着年龄的增长,斑点、毛细血管扩张和毛孔面积的得分增加,而皮脂含量、卟啉、水合作用和皮肤弹性的得分则下降(图 1B),这与我们对皮肤老化的认识一致。此外,随着年龄的增长,皮肤颜色变得更加暗淡(L*)、偏黄(b*)和偏红(a*),这与之前的研究结果一致(图 1B)。值得注意的是,pH 值与年龄无明显关系,而反映皮肤屏障功能的经表皮失水率(TEWL)却意外地随着年龄的增长而下降。这些不一致可能进一步意味着皮肤老化与实际年龄存在偏差。在面部的三个部位,这些关联通常具有高度的一致性。
然后,我们使用置换多元方差分析(PERMANOVA)评估了与不同宿主特征相关的皮肤微生物变化。有 14 个因素与皮肤微生物群明显相关(校正后 p < 0.05,图 1C)。总体而言,卟啉对微生物群变异的解释最大,尤其是在细菌群落中,这与之前在北美志愿者中的发现一致。其次是年龄和水合水平。值得注意的是,在真菌群落中,年龄对变异的影响很小,而性别对变异的影响却很显著(图 1C)。值得注意的是,许多皮肤表型(如肤色(L*、a*)、毛细血管扩张、pH 值和 TEWL)对真菌群落变异的解释要多于细菌群落。病毒群落通常被认为是皮肤微生物群的短暂成员,其变异性也与卟啉水平、年龄和其他皮肤特征有关(校正后 p < 0.05,图 1C)。协惯量分析显示病毒群落和细菌群落之间存在显著关系(置换检验,p < 0.01),细菌的丰度与相应的噬菌体高度相关(图 S2A,B),这表明跨界的微生物之间的相互作用是生态系统群落平衡的基础。
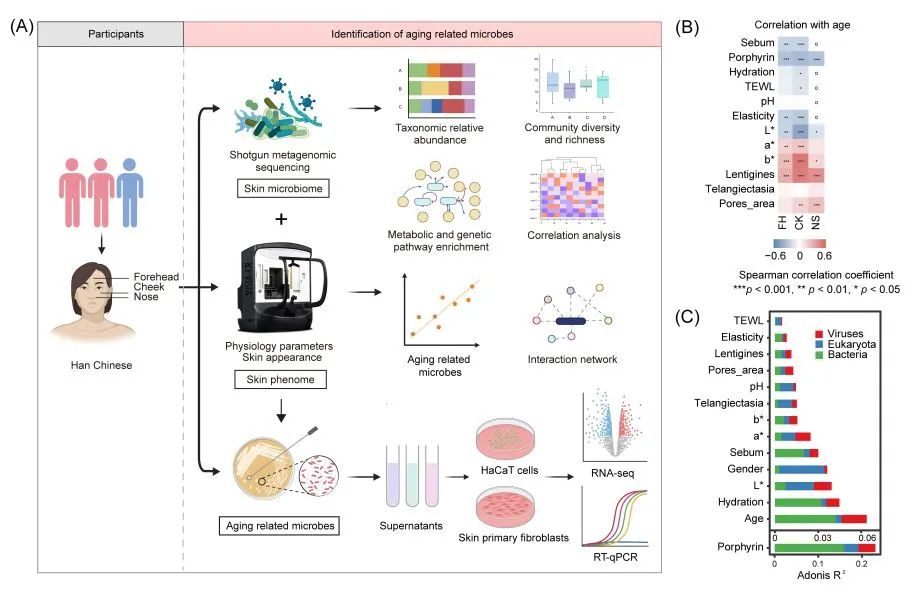
图1. 研究流程、相关性分析和微生物组成的影响
(A) 本研究的整体工作流程;(B) 表型参数与年龄之间的斯皮尔曼相关性。FH:前额;CK:脸颊;NS,鼻旁。L* = 皮肤暗度/亮度;a* = 红斑;b* = 晒黑。L* 值的增加表示从黑色向白色过渡,而 a* 值则从绿色向红色过渡,b* 值则从蓝色向黄色过渡。蓝色表示负相关。“o "表示由于鼻旁解剖学限制而缺少经表皮失水(TEWL)、水合作用、皮脂、pH 值和弹性方面的数据。颜色条代表相关值。斯皮尔曼相关性的显著性水平为: *,p < 0.05;**,p < 0.01;***,p < 0.001;(C) 柱状图说明了各变量对细菌、真菌和病毒组成的影响大小。图中显示了对物种有显著影响的变量。校正后的 R2 值代表变量解释的微生物组成变化的比例。不同颜色代表不同界别。部分用 BioRender.com 制作。
与年龄有关的皮肤微生物组动态
除卟啉外,年龄是影响细菌变异的最大因素(图 1C)。为了确定与年龄相关的物种,我们探讨了不同年龄组的细菌动态:年轻组(20-35 岁,74 人)、中年组(36-50 岁,131 人)和老年组(50 岁以上,89 人)(图 2A)。
总体而言,所有受测面部部位的细菌多样性随着年龄的增长而增加(图 2B),这与之前的研究结果一致。此外,各组之间的β多样性显示,年龄差异越大,皮肤细菌微生物组的变化越显著(图 2C)。基于距离的冗余分析表明,不同年龄组的微生物物种之间存在明显的差异(图 2D)。通过比较不同年龄组的细菌种类,发现了 58 种与年龄相关的细菌种类(图 2E)。虽然大多数细菌种类随着年龄的增长而增加,特别是莫拉氏菌属(主要是Moraxella osloensis)、金黄杆菌属Chryseobacterium spp.(主要是Chryseobacterium taklimakanense)、伊丽莎白菌属Elizabethkingia spp.和副球菌属Paracoccus spp.,但有两种细菌种类随着年龄的增长而逐渐减少,即痤疮丙酸杆菌Cutibacterium acnes和气微菌Aeromicrobium choanae(图 2E)。在其他研究中,A. choanae与皮肤微生物有关。我们发现该物种的丰度很低,但同属的另一个物种 Aeromicrobium phoceense 已从皮肤中分离出来。值得注意的是,痤疮丙酸杆菌Cutibacterium acnes和气微菌Aeromicrobium choanae显示出一种协同效应,彼此呈正相关,而与其他年龄增加的物种呈负相关(图 2F)。除皮肤细菌外,我们还发现了三种与年龄相关的病毒(Spearman's p < 0.05):Betapapillomavirus 3、Staphylococcus virus phiETA和Streptococcus phage IPP18(图 S2C)。

图2. 细菌群落在衰老过程中的动态变化
(A) 不同年龄组:年轻组(20至35岁,N = 74),中年组(36至50岁,N = 131),老年组(50岁以上,N = 89);(B) 在衰老过程中,细菌群落的α多样性略有增加。使用Wilcox检验确定显著性;(C) 三个年龄组之间细菌群落的β多样性存在差异。使用Wilcox检验确定显著性;(D) 基于距离的冗余分析(dbRDA)展示了不同年龄组之间细菌群落组成的差异。每个点代表一个样本。颜色代表年龄组。形状代表不同部位。线条代表不同年龄;(E) 细菌类群与年龄之间的关联。颜色代表三个部位:绿色为CK,橙色为FH,紫色为NS;(F) 与年龄相关的物种共现网络。节点代表与年龄相关的物种。节点颜色代表门(phylum)。红色线条表示正相关,蓝色线条表示负相关。
皮肤微生物组与皮肤老化特征高度相关
皮肤老化是一个动态过程,涉及外观和生理参数的一系列表型变化。这些表型可以反映生态位条件,如 pH 值和水合作用,或者是微生物代谢活动的最终表现。
为了明确与表型相关的微生物动态,我们在微生物组和衰老表型之间进行了跨界的斯皮尔曼相关性分析(图 3)。在 20 种最丰富的细菌中,痤疮丙酸杆菌C. acnes和奥斯陆莫拉菌M. osloensis呈现出相反的趋势。C. acnes的丰度与卟啉、皮脂、TEWL、水合作用和毛孔面积的水平呈正相关,但与年龄和肤色(b*)呈负相关。相比之下,M. osloensis 容易出现衰老特征,如水合作用降低、色斑增加和肤色变黄(图 3A)。另一种众所周知的皮肤共生菌表皮葡萄球菌Staphylococcus epidermidis也表现出与C. acnes相关的类似趋势;然而,除了 TEWL 外,其他相关性并不显著(图 3A,图 S3)。
虽然真菌在微生物群落中只占少数,但其细胞大小通常是细菌细胞的 100 倍,而且许多真菌通过其独特的新陈代谢被认为对宿主表型的形成具有影响力。因此,我们进一步评估了与皮肤表型相关的真菌成员。许多真菌物种与独特的皮肤特征相关(图 3B)。值得注意的是,许多马拉色菌Malassezia与皮肤皮脂水平呈正相关,这与它们的亲脂特性是一致的。
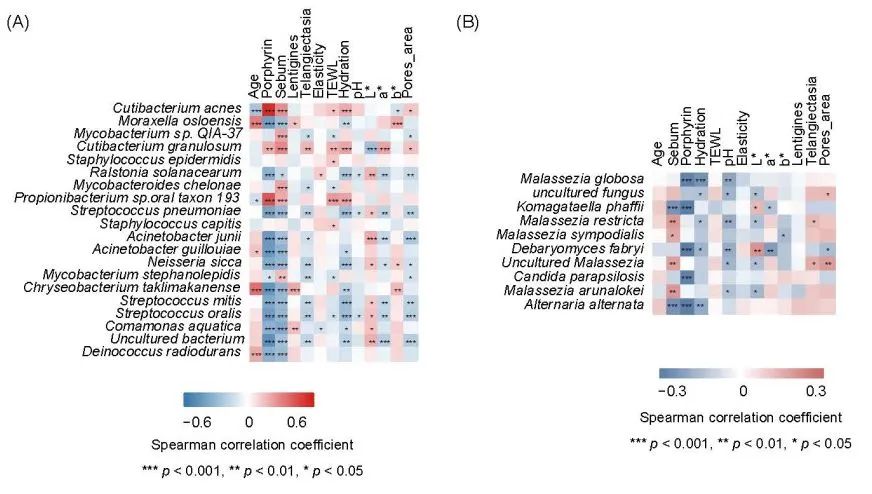
图3. 细菌、真菌与表型参数之间的相关性
热图展示了前额皮肤中前20种细菌 (A) 和前10种真菌 (B) 的丰度与皮肤表型参数之间的Spearman相关性。蓝色表示负相关;红色表示正相关。Spearman相关性中的显著性水平为: *,p < 0.05;**,p < 0.01;***,p < 0.001。
基于机器学习的皮肤年龄建模,定位早衰/延迟衰老相关微生物物种
虽然时序年龄是人口中大多数皮肤老化表型的核心,但一个人的感知年龄(或称 “肤龄”)可能会偏离其时序年龄。我们开发了一种基于机器学习的应用皮肤年龄算法来评估感知年龄。该算法在 200,000 张图像上进行了训练,使用时序年龄作为训练标签,并在感知年龄群组(N = 5,768 人)中进行了微调。模型预测值与调查人员感知的平均面部年龄之间的相关系数达到了 0.97。
采用多元线性回归(MLR)模型预测皮肤年龄。使用MLR模型配置得出的残差标准误差(RAE)为 3.639 岁,R2 为 0.7665。为了确定与衰老/抗衰老相关的微生物种类,我们筛选了Δ年龄(预测年龄与时序年龄之差)大于RAE的个体,并将其进一步分为早衰组(n = 30)和延迟衰老组(n = 27)(图4A)。我们依次比较了两组之间皮肤微生物组成的差异。早衰组的细菌种类更多,如奥斯陆莫拉菌M. osloensis、凝结芽孢杆菌Chryseobacterium greenlandense、格陵兰金黄杆菌Chryseobacterium greenlandense和施氏假单胞菌Pseudomonas thivervalensis。相比之下,痤疮丙酸杆菌C. acnes在延迟衰老组中含量较高(图 4B)。为了探索衰老相关物种的潜在功能,我们比较了不同组间的细菌 KEGG KOs。值得注意的是,早衰组的微生物组在双组分调控系统、抗性和药物外排转运体/泵方面表现出显著的基因模块富集(图 4C)。耐药性基因的富集可能是由于频繁或长期接触抗生素,这表明皮肤微生物组经历了选择性压力,导致耐抗生素菌株的增加。双组分系统是细菌中的信号转导系统,可对适应性环境变化(如 pH 值、温度或抗生素的存在)做出反应。早衰组微生物群中这些功能的丰富表明,皮肤微生物群可能需要更频繁地调整其生理状态,以适应更恶劣或不断变化的微环境。相比之下,延迟衰老组的微生物组富含负责运输碳水化合物的磷转移酶系统和碳水化合物代谢中心,这可能代表了这些共生菌的活跃生长和能量需求。
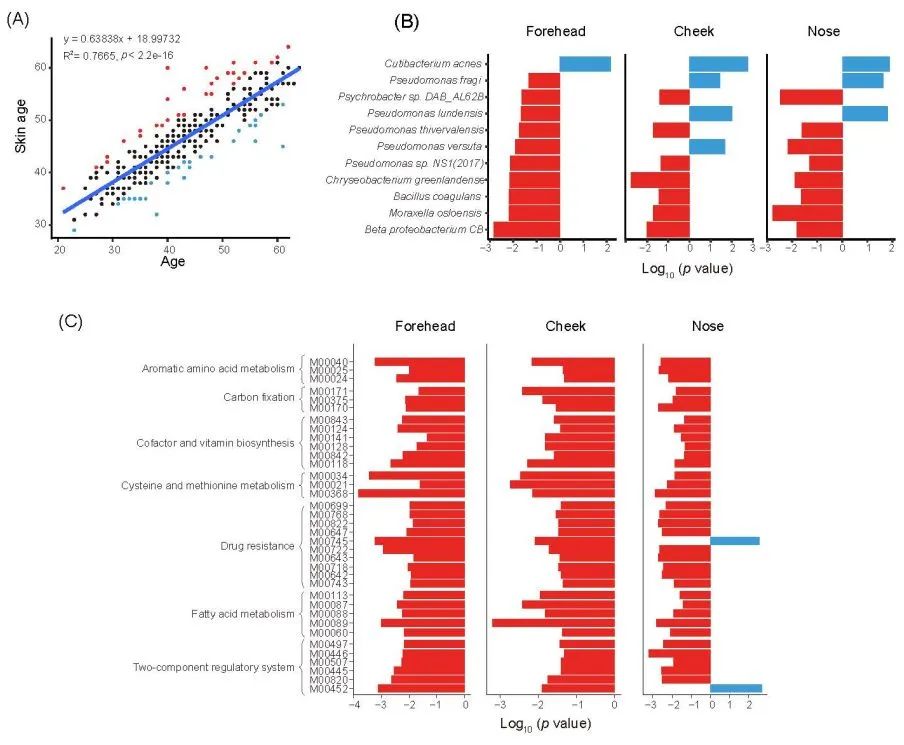
图4. 早衰组与延缓衰老组之间微生物组成和功能的差异
(A) 使用多元线性回归 (MLR) 模型预测皮肤年龄。红色表示属于早衰组;蓝色表示属于延缓衰老组;(B) FH、CK 和 NS 部位年轻皮肤组与老年皮肤组之间的微生物组成差异。红色表示在老年皮肤组中富集的细菌;蓝色表示在年轻皮肤组中富集的细菌;(C) FH、CK 和 NS 部位早衰组与延缓衰老组之间细菌功能特征的差异。柱状图展示了差异性的KEGG模块的相对丰度。红色表示在早衰组中富集的模块;蓝色表示在延缓衰老组中富集的模块。
奥斯陆莫拉菌调节人角质细胞和成纤维细胞的胶原代谢和细胞外基质组装
奥斯陆莫拉菌M. osloensis和痤疮丙酸杆菌C. acnes也被称为中国人皮肤中的皮肤型决定性物种,它们与年龄和皮肤老化表型显著相关(图 2E、3A 和 4B)。痤疮丙酸杆菌C. acnes与年龄和衰老特征呈负相关,如皮脂/卟啉生成增加、弹性和水合水平提高、色斑减少等,而奥斯陆莫拉菌M. osloensis则表现出相反的相关性,容易出现衰老特征。我们还评估了表型与表皮葡萄球菌S. epidermidis(一种著名的优势皮肤共生菌)的相关性,发现表皮葡萄球菌与皮肤表型的相关性很弱(图 S3)。
皮肤特征与细菌之间的相关性可能与皮肤生态位驱动的细菌变异有关,反之亦然。为了进一步探索这些关联背后潜在的宿主-微生物相互作用,我们用上述共生菌的上清液对人类表皮角质细胞(HaCaT)和原代真皮成纤维细胞进行了 RNA-seq 分析(图 5A)。对 HaCaT 细胞进行的基因本体(GO)富集分析表明,经 M. osloensis 条件培养基处理后,调控胶原降解过程、细胞外基质解体和胶原代谢过程的差异表达基因(DEGs)明显富集(图 5B)。定量实时聚合酶链式反应(RT-qPCR)结果证实,M. osloensis 上清液处理组中多种基质金属蛋白酶(MMPs)的 RNA 表达显著上调(图 5C、5D),这与 MMPs 与皮肤老化之间的关系一致,因为众所周知,这些酶是胶原降解和皮肤起皱的主要因素。根据细胞衰老数据库 Senequest (http://Senequest.net),当人类真皮成纤维细胞与 M. osloensis 的上清液(而不是 C. acnes 或 S. epidermidis)一起培养时,结果还显示了与细胞衰老有关的 DEGs 的显著积累。在M. osloensis处理的条件培养基中,成纤维细胞的基因表达发生了变化,表现为细胞周期停滞、衰老相关分泌表型、大分子损伤以及失调的代谢特征(图 5E)。这些细胞变化可能会对组织微环境产生有害影响,并导致多种与年龄相关的病症。我们发现,S. epidermidis和 C. acnes诱导的 MMP 表达量不如M. osloensis诱导的显著,尤其是在原代真皮成纤维细胞中。这表明M. osloensis在影响皮肤细胞模型中的胶原代谢和细胞外基质组装方面具有潜在的独特作用。
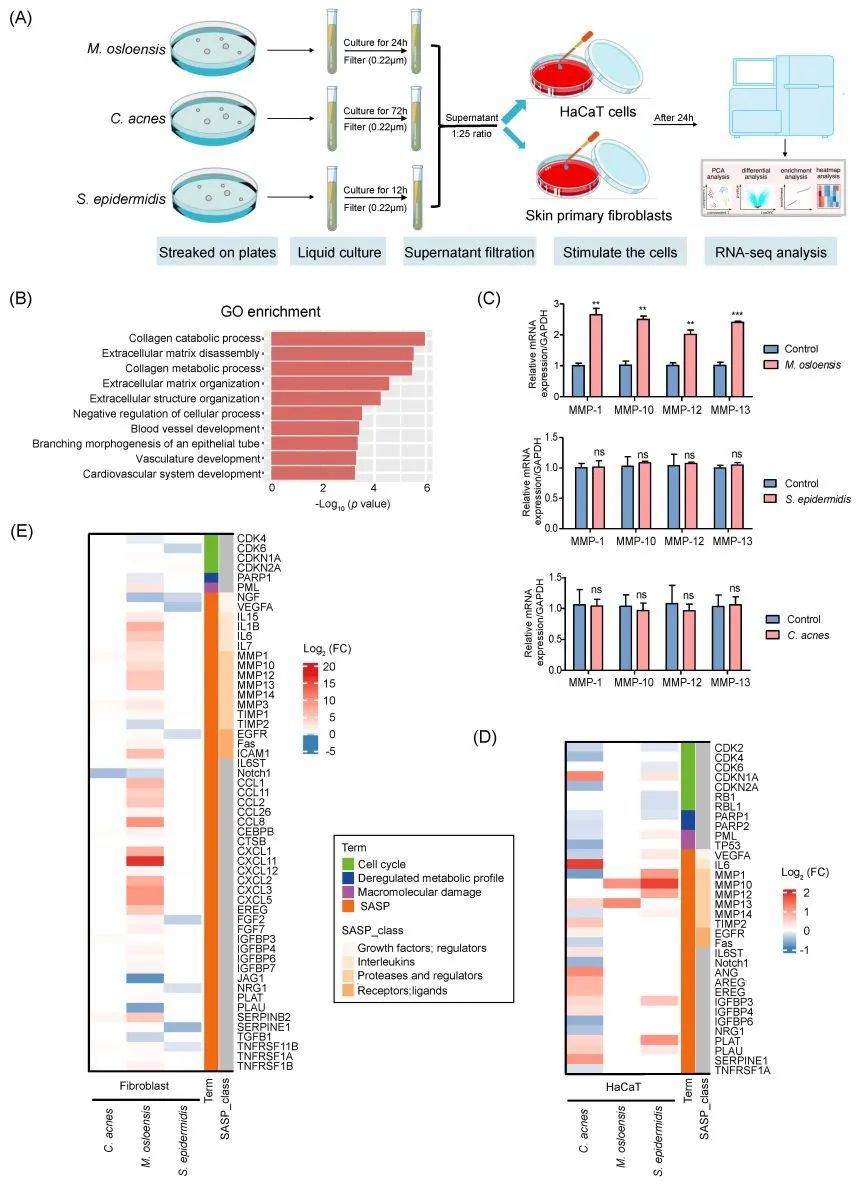
图5. 与三种皮肤细菌共培养的角质形成细胞和成纤维细胞的基因表达情况
(A) 皮肤细胞被细菌上清液刺激的示意图。对HaCaT细胞和原代真皮成纤维细胞进行RNA-seq分析,这些细胞在用M. osloensis、C. acnes和S. epidermidis的上清液(分别在R2A、RCM或LB培养基中培养,OD600 0.8-1.0,n = 3)刺激24小时后进行分析。以细菌培养基(R2A、RCM或LB)刺激的细胞作为对照组;(B) 基因本体(GO)富集分析显示了用M. osloensis的细菌上清液处理的HaCaT细胞中前10个富集通路(p < 0.05);(C) 通过定量PCR(qPCR)分析M. osloensis、C. acnes和S. epidermidis的上清液分别刺激HaCaT细胞24小时后几种基质金属蛋白酶(MMPs)的基因表达(*,p < 0.05;**,p < 0.01;***,p < 0.001,数据汇总自n = 3次独立实验);(D)和(E)为三个微生物物种的上清液分别处理过的HaCaT细胞(D)和成纤维细胞(E)中的衰老相关基因表达变化的热图(校正后的p < 0.05)。灰色条表示相应的基因没有进一步的子分类。
与年龄有关的皮肤真菌动态
通过鸟枪法宏基因组数据,我们可以研究真菌群落的动态变化。与对细菌群落的重大影响相比,年龄对真菌群落变化的影响较小(图 1C)。在年龄与真菌香农指数之间没有发现明显的相关性(p = 0.09)(图 6A)。然后,我们探讨了不同年龄组真菌的动态变化。老年组的真菌 α 多样性高于年轻组(图 6B)。这与之前使用内部转录间隔 1(ITS1)基因扩增片段测序法对撒丁岛 65 人进行的研究和对韩国健康女性进行的研究一致。基于Bray–Curtis距离的主坐标分析(PCoA)显示,按年龄对真菌群落进行分组具有统计学意义(p = 0.001)(图 6C)。Kruskal-Wallis 检验显示,年轻和中年组真菌群落组成差异明显大于老年组(p < 0.05)(图 6D)。
为了确定与年龄相关的真菌分类群,进行了 Wilcoxon 秩和检验(图 6E)。有 11 种真菌在年轻组明显富集,32 种在老年组明显富集。例如,球形马拉色菌Malassezia globosa和法布里德巴利酵母Debaryomyces fabryi在年轻组中富集。随后,我们进行了斯皮尔曼相关分析,以研究差异真菌与皮肤特征之间的关系(图 6F)。结果显示,大多数差异真菌与年龄呈正相关,而与卟啉和皮脂水平呈负相关。
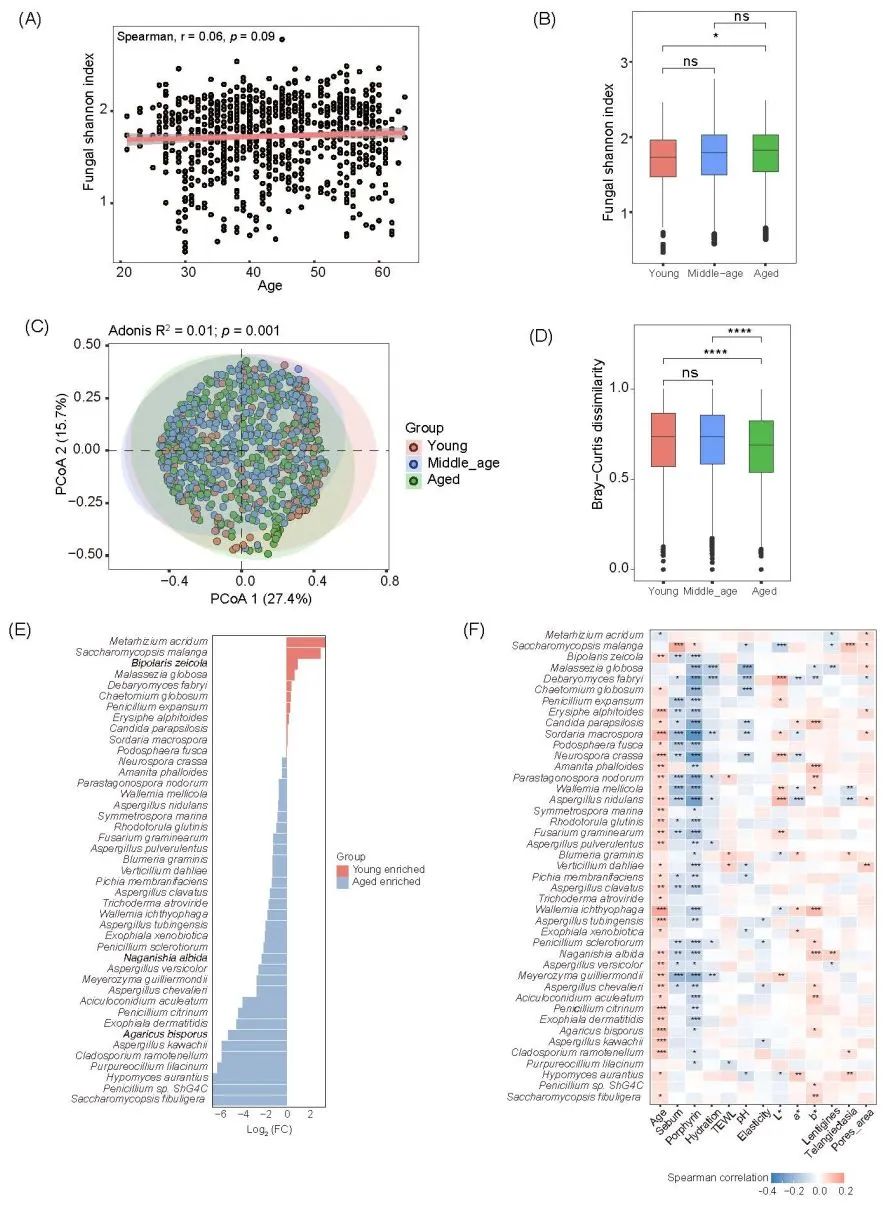
图6. 三个年龄组中真菌群落的动态变化
(A) 真菌群落中α多样性与年龄的Spearman相关性分析;(B) 箱线图比较了年轻组(20−35岁,N = 74)、中年组(36−50岁,N = 131)和老年组(> 50岁,N = 89)之间真菌α多样性的差异。使用Wilcoxon检验确定显著性;(C) 基于Bray–Curtis距离的主坐标分析(PCoA)显示了不同年龄组中每个个体的真菌群落在物种水平上的分布。每个年龄组内的样本通过95%置信椭圆聚类。使用PERMANOVA检验确定显著性;(D) 三个年龄组之间真菌群落的β多样性。通过物种水平上两个样本之间的Bray-Curtis距离来测量群落组成的相似性;(E) 使用Wilcoxon秩和检验确定年轻组和老年组之间真菌组成的差异。红色柱状条表示在年轻组中富集的真菌,蓝色柱状条表示在老年组中富集的真菌;(F) 热图展示了(E)中差异真菌与13个宿主因素之间的Spearman相关性分析。统计显著性如下表示:*,p < 0.05;**,p < 0.01;***,p < 0.001。
讨 论
建立各种人类共生系统背后的驱动力仍不清楚。它是一个随机过程还是涉及选择,或者是哪种选择(如果有的话),这些都是该领域最令人担忧的问题。以往的研究表明,宿主的中性漂移和生态位选择可导致人类微生物群的生物多样性和生物地理学。有研究认为,不同的皮肤生态位会影响当地微生物的组成。反过来,皮肤共生菌能够通过将宿主的营养物质转化为酸性代谢物来改变局部的生理机能,如 pH 值或水合作用 。
值得注意的是,在本研究中,我们发现在所有测量变量中,卟啉对微生物变异性的解释率最高,这与最近一项针对北美人群的大型队列研究结果一致。这些结果有力地表明,无论在哪个种族的人群中,产生卟啉的物种都在维持面部皮肤生态系统的稳定性方面发挥着核心作用。Cutibacterium granulosum、Cutibacterium avidum和 Cutibacterium modestum(以前称为 “Propionibacterium humerusii”)等几种丙酸杆菌物种都能产生卟啉;然而痤疮丙酸杆菌C. acnes是主要的卟啉贡献者。痤疮丙酸杆菌是人体皮肤中最重要的共生微生物之一,能促进皮肤中油酸的积累,并将脂肪酸或其他油脂成分代谢为丙酸和乙酸,从而消除有害微生物,保护人体皮肤。该物种还倾向于生活在皮脂丰富的环境中,生物多样性较低。鉴于这些结果,我们认为痤疮丙酸杆菌是皮肤生态系统中的基础物种,它控制着相关物种的生物多样性,调节着关键的生态系统过程,通常具有重要的文化价值和共鸣。
皮肤老化与皮肤生理变化有关,包括与皮肤微生物群落的相互作用。许多 16S rRNA 基因调查报告了年龄对不同种族人群皮肤微生物群组成的影响。尽管不同人群的物种差异很大,但在亚洲人和西欧人中都观察到老年人的细菌α多样性/物种丰富度较高。具体而言,老年人群中的棒状杆菌Corynebacterium数量大幅增加,但丙酸杆菌Cutibacterium数量减少。这一结果与以下事实相矛盾:生物多样性越高,生态稳定性越好(皮肤越健康),而生物多样性降低往往与脆弱的稳定性和皮肤状况有关。由于老年人要忍受更多的皮肤问题,我们认为老年人多样性的增加是生态系统中基础物种(即痤疮丙酸杆菌)减少的被动结果。这也与皮肤皮脂和卟啉水平在衰老过程中下降的事实相吻合,痤疮丙酸杆菌的减少可以照顾到其他物种的生存。
在这项研究中,我们发现痤疮丙酸杆菌与年龄和皮肤老化表型显著相关(图 2E、3A 和 4B)。痤疮丙酸杆菌与年龄和衰老特征呈负相关,如皮脂/卟啉生成增加、弹性和水合水平提高以及色斑减少。Larson 等人同样发现痤疮丙酸杆菌在年轻成人皮肤中含量较多,而老年人(年龄≥ 65 岁)皮肤中的痤疮丙酸杆菌则大量减少。老年人中痤疮丙酸杆菌的减少可能是由于与年龄有关的皮肤生理变化,如皮脂分泌减少。痤疮丙酸杆菌的减少可能为机会性微生物的定植和繁殖创造了适宜的环境,从而导致微生物群落更加多样化。与年轻人相比,老年人皮肤生态位的选择性较低,导致不稳定性增加,免疫或营养选择性丧失。此外,Larson 等人还发现,痤疮丙酸杆菌的相对丰度与脸部、躯干、肘前窝和手部的洛克伍德虚弱指数(Rockwood Frailty Index)显著相关。这一结果促使我们思考痤疮丙酸杆菌是否在人类衰老、虚弱和与衰老相关的疾病中扮演重要角色,这需要我们在未来进行进一步研究。
一系列研究还提出了其他微生物组与表型之间的关联。例如,一项关于韩国人皮肤微生物组的研究发现,劳森氏菌Lawsonella与皮肤水分和黄褐斑呈负相关,葡萄球菌和棒状杆菌与紫外线斑数量呈负相关,而与 TEWL 呈正相关,金黄色葡萄球菌与皮肤水分参数呈负相关。然而,这些研究大多需要功能验证的直接证据。在本研究中,我们对与衰老相关的物种进行了特征描述,如 M. osloensis。对这一物种的研究很少,只有极少数报告指出它是免疫力低下的成年人的病原体。然而,该物种已被证明是人类皮肤共生菌的重要成员,并已成为汉族人皮肤上第二丰富的物种。在这里,我们证实了 M. osloensis 与早衰特征的关联,并进一步验证了其促进皮肤细胞衰老的能力,这也解释了中国人和其他老年人群的早衰表型 。
尽管在解密人类病毒群方面取得了快速进展,但仍有一些障碍有待充分表征和利用。由于方法上的困难,人类皮肤病毒组是皮肤微生物群中已知最少的群落。病毒究竟是人体皮肤菌群的正常组成部分,还是与宿主互利互惠,仍有待确定。我们的研究队列中发现了大量噬菌体,这与之前的一项研究结果类似,该研究报告称北美人群中存在大量核心噬菌体。值得注意的是,噬菌体的丰度与相应细菌的丰度高度相关。我们的数据进一步揭示了表型组与卟啉、年龄、肤色、毛孔面积和色斑等皮肤特征的潜在相互作用,这在一定程度上解释了病毒组的变异性。鉴于病毒的低丰度和对肠道微生物组的了解,这些病毒组与表型的关联很可能是病毒与细菌相互作用的结果。
我们的研究侧重于面部微生物组,特别分析了前额、脸颊和鼻旁的样本,这些部位受环境因素的影响最大,也是明显衰老的标志。虽然这种方法为了解皮肤微生物群随着年龄的增长而发生的变化提供了重要依据,但我们也承认,它所展示的观点仅限于面部皮肤。不同的身体部位有其独特的微环境,可能会对衰老表现出不同的反应。因此,我们的研究结果虽然重要,但可能并不能完全代表整个皮肤微生物组的系统性变化。未来的研究应该考虑更大的样本,以便更全面地了解皮肤微生物组在整个衰老过程中的演变。
在本研究中,我们展示了体外实验的数据,这些数据表明M. osloensis在调节 MMPs 的表达和潜在影响皮肤老化方面发挥作用。尽管这些发现提供了宝贵的见解,但我们认识到体外研究固有的局限性,以及在更复杂的生物系统中验证这些结果的重要性。通过使用动物模型,我们可以在一个多种细胞类型和系统因素相互作用的综合系统中观察 M. osloensis 的作用,这无疑会加强我们的主张的证据。这种模型能更有代表性地描述皮肤老化过程和微生物组在生物体内的作用。然而,由于我们的研究设计和可用资源的限制,动物模型实验并没有包括在本研究中。尽管存在这种限制,但我们相信,我们的体外研究结果为今后的研究奠定了坚实的基础。我们强调了体内实验的重要性,以证实并扩展我们对M. osloensis和其他皮肤微生物群如何促进皮肤老化复杂机制的理解。
结 论
在本研究中,通过对鸟枪法宏基因组测序结果的解释,并辅以依赖培养的方法,我们更好地了解了人类皮肤微生物群落的组成、多样性和变异性,甚至提供了一些有关功能和机理属性的相关信息。在本研究中,我们(i) 分析了与年龄/表型相关的皮肤微生物组动态,(ii) 预测了与早衰/延迟衰老相关的微生物物种,主要是 M.osloensis 和 C. acnes,(iii) 在体外验证了一些宿主-微生物相互作用的生物学功能。
总之,目前的研究已经超越了皮肤上存在哪些微生物的问题,而是评估它们的功能与对皮肤影响之间的联系。我们系统地描述了皮肤微生物组-表型组关联网络,尤其是衰老表型。此外,我们还通过体外功能实验验证了一些预测的关联,因为深入了解衰老过程的分子通路对于寻找针对皮肤微生物群的产品以改善皮肤表型至关重要。因此,我们的研究对于设计针对各种年龄相关的皮肤状况的干预策略非常重要。
代码和数据可用性
本研究的测序数据已存储在CNGBdb的CNSA(https://db.cngb.org/cnsa/),获取号为CNP0000635,并存储在NODE(https://www.biosino.org/node/index),获取号为OEP001168。为了更好地展示基因目录的注释信息,并为有兴趣使用我们的数据集和下载特定数据集的研究人员提供指导,我们建立了一个网站(https://db.cngb.org/microbiome/genecatalog/genecatalog/?gene_name=Human%20Skin%20(10.9M))。数据和脚本保存在https://github.com/ZQ19961021/skin-microbiome.git。
引文格式:
Jing-jing Xia, Qian Zhong, Zhi-ming Li, Qing-zhen Wei, Liu-yi-qi Jiang, Cheng Duan, Hui-jue Jia, Yi-mei Tan, Lian-yi Han, Jean Krutmann, Jiu-cun Wang, Xiao Liu. 2024. Culture-dependent and -independent approaches reveal the role of specific bacteria in human skin aging. iMetaOmics e26. https://doi.org/10.1002/imo2.26
作者简介

夏晶晶(第一作者)
● 毕业于华中科技大学同济医学院临床医学六年制、德国马普所心肺所博士(summa cum laude)、德国科隆大学医学院皮肤科博士后。
● 2016年起师从德国科学院院士Jean Krutmann教授、复旦大学生科院王久存教授,协助中德导师组建“环境与皮肤”实验室。现任粤港澳大湾区精准医学研究院(广州)微生物与免疫研究中心青年研究员。主要研究方向为皮肤微生物组与皮肤屏障、外源性皮肤衰老等。成果发表在Developmental Cell、Microbiome、Journal of Investigative Dermatology、Advanced Science等期刊。

钟倩(第一作者)
● 复旦大学生命科学学院遗传学博士。
● 研究方向集中于探索皮肤微生物-表型互作、微生物物种间互作关系,相关学术成果已发表于iMetaOmics、Phenomics、Clinical Rheumatology等期刊。

李志明(第一作者)
● 复旦大学生命科学学院生物与医药博士。
● 研究方向为宏基因组和多组学,已在Microbiome、Advanced Science、Molecular psychiatry、Aging cell等期刊发表学术论文。

魏情珍(第一作者)
● 复旦大学生命科学学院遗传学在读博士。
● 相关学术成果已发表于iMetaOmics、Frontiers in Microbiology等期刊。

刘晓(通讯作者)
● 研究员,清华大学深圳国际研究生院副教授,博导。
● 南京大学本科,哥本哈根大学生物学博士。曾领导和参与多项基因组学重大项目合作,包括1000 genomes, ICGC, ACRG等。在Nature Genetics、Science、Genome Research等国际学术期刊发表SCI收录科研论文90余篇,其中第一和通讯作者论文40多篇,总引用超过30000次, H指数44 (Google学术)。担任包括Nature Communications, Cell Research等十多家SCI期刊编委或审稿人,授权国内外专利20余项。主持和参与多项科技部和地方科研项目。现为美国免疫学会和国际免疫联合会会员,深圳市海外高层次人才B类,国家科技部在库专家。目前主要研究方向是人类系统免疫学和细胞免疫工程、免疫组学等。

王久存(通讯作者)
● 复旦大学生命科学学院教授,复旦大学人类遗传学与人类学系系主任,重点研发计划首席科学家。
● 研究方向为硬皮病等风湿免疫性疾病的遗传学和分子机制研究及皮肤表型组学研究。近年以通讯作者在Blood、Annals of the Rheumatic Diseases、National Science Review、Microbiome等期刊发表研究成果。承担国家重点研发计划、国家自然科学基金重点项目、中国医学科学院创新基金、上海市市级科技重大专项子项目等。

Jean Krutmann(通讯作者)
● 德国科学院院士、IUF莱布尼茨环境医学研究所所长、国际著名皮肤学专家。
● 研究领域为皮肤毒理学、免疫皮肤病学和光皮肤病学,重点关注环境引起的皮肤疾病和皮肤老化。发表学术论文600余篇,专著10余部。美国皮肤病研究学会(SID)、日本皮肤病研究学会(JSID)荣誉会员,曾获多个重要国际奖项,如Arnold Rikli 奖、Albrecht Fleckenstein奖、Paul Gerson Unna 奖、Oscar Gans奖、CE.R.I.E.S.研究支持奖、Dermopharmacy创新奖、Tanioku Kihei 纪念奖等。
iMetaOmics
更多资讯
● iMeta姊妹刊iMetaOmics(定位IF>10)欢迎投稿!(2024.2.27)
● iMeta姊妹刊iMetaOmics编委招募 (定位IF>10) (2024.3.2)
● iMeta姊妹刊iMetaOmics电子版和印刷版ISSN申请获批(2024.4.1)
● iMeta姊妹刊iMetaOmics投稿系统正式上线(2024.4.17)
● iMeta姊妹刊iMetaOmics主编正式官宣(2024.4.22)
● 出版社iMetaOmics主页正式上线!(2024.4.28)
● iMetaOmics | 浙江大学宗鑫组揭示两猪种宿主-肠道菌群互作差异
● iMetaOmics | 罗鹏/袁硕峰/苗凯/程全发表STAGER: 生成式人工智能可靠性的标准化测试和评估推荐
● iMetaOmics | 徐州医科大杨欢组揭秘沙门氏菌-宿主-微生物群在免疫与代谢中的相互作
● iMetaOmics | 中科院动物所金坚石组综述16S rRNA基因扩增子测序技术的“前世今生”
● iMetaOmics | 浙大张天真组完成二倍体棉种泛基因组构建
● iMetaOmics | 张勇/李福平-先进糖蛋白组学在男性生殖研究中的潜在应用
● iMetaOmics | 暨南大学潘永勤/杨华组-炎症蛋白联合检测利于诊断甲状腺乳头状癌和结节性甲状腺肿
● iMetaOmics | 张开春组利用多组学方法揭示甜樱桃加倍后果色变化的候选基因
● iMetaOmics | 杜娟/林婷婷-慢性泪囊炎患者眼部菌群类型和纵向菌群变化
● iMetaOmics | 陈汉清/陈俊综述有关肝细胞癌治疗的新兴纳米医学策略
● iMetaOmics | 基因组所刘永鑫/卢洪评述微生物在提高杂种优势中的作用
● iMetaOmics | 上科大刘雪松组开发基于通路的肿瘤细胞鉴别工具TCfinder
● iMetaOmics | 中山大学刘鹏/邹宇田-整合人工智能实现HER2阳性乳腺癌精准管理
● iMetaOmics | 安徽农大李晓玉组-丛枝菌根真菌对玉米内生菌群的影响
● iMetaOmics | 徐涛/黄蓉/苏国海-急性冠脉综合征纵向多组学队列建设
● iMetaOmics | 通过整合宏组学促进人类与环境健康发展
●iMetaOmics | 苏州大学林俊组-揭示活性微生物及益生元/益生菌与关节炎联系
更多推荐
(▼ 点击跳转)
iMeta | 引用13000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 德国国家肿瘤中心顾祖光发表复杂热图(ComplexHeatmap)可视化方法

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

2卷2期封底

2卷4期封底

3卷2期

3卷3期

3卷3期封底

3卷4期

3卷4期封底

1卷1期
期刊简介
“iMeta” 是由威立、肠菌分会和本领域数百千华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括视频投稿、可重复分析、图片打磨、青年编委、前3年免出版费、50万用户的社交媒体宣传等。2022年2月正式创刊发行!发行后相继被Google Scholar、ESCI、PubMed、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.7,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,同学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 727
727

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









