






微生物组分析方法与功能挖掘
高云云1 杨海飞1, 2 吕虎杰1 刘永鑫1
(1. 中国农业科学院深圳农业基因组研究所,深圳 518120;2. 青岛农业大学生命科学学院,青岛 266109)
高云云, 杨海飞, 吕虎杰, 刘永鑫. 2024. 微生物组分析方法与功能挖掘. 生物技术通报 40: 98-107.
https://biotech.aiijournal.com/CN/10.13560/j.cnki.biotech.bull.1985.2024-0788
摘 要 :微生物是生命科学研究中不可或缺的重要资源,其研究对推动科学进步、促进人类健康和改善环境质量等具有重要意义。随着二、三代高通量测序技术的迅猛发展,我们对微生物世界的认知得到了极大提升,在面对大量的微生物组数据时,选择适当的分析方法以实现快速、准确的信息挖掘显得尤为关键。本文对近年来微生物组研究的进展进行了系统的回顾与分析,着重更新了扩增子、培养组和宏基因组等二代短读序宏基因组数据的分析工具,纳入了三代长读序宏基因数据的处理方案,并提出了标准化数据分析流程的必要性。此外,本文结合解析微生物组的实例案例,侧重介绍其在植物与根系微生物互作、微生物多样性等方面的应用实例,探讨了不同方法在微生物组构成、结构和功能分析中的优劣,展示了宏基因组数据挖掘在应用方面的潜力,以期拓宽宏基因组数据挖掘的研究思路。最后,本文指出了当前微生物组研究中的不足和面临的挑战,并展望了未来微生物组研究技术的标准化与流程化方面的发展趋势,以期加速微生物组的功能与应用的研究进程。
关键词:微生物组;宏基因组;扩增子;分析方法;功能挖掘
DOI: 10.13560/j.cnki.biotech.bull.1985.2024-0788
Analytical Approaches and Functional Insights for Microbiome Studies
GAO Yun-yun1 YANG Hai-fei1, 2 LYU Hu-jie1 LIU Yong-xin1
(1. Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen518120; 2. College of Life Sciences, Qingdao Agricultural University, Qingdao 266109)
Abstract:Microbiomes are crucial resources in life science, playing vital roles in advancing scientific knowledge, promoting human health, and improving environmental quality. The rapid evolution of second- and third-generation metagenomic technologies has greatly enhanced our understanding of microbial world. With the proliferation of microbiome data, the need to select appropriate analytical methods for efficiently extracting information has become increasingly essential. Here, we provide a comprehensive review of recent progress in microbiome studies, focusing on updated analytical tools for short-read second-generation sequencing data, including amplicon, culture-based methods, and metagenomic data. In addition, we offer the processing strategies for long-read third-generation sequencing data. We highlight the necessity for standardized data analysis workflows and present case studies that demonstrate the application of these methods in exploring microbiome interactions, particularly in plant and root-associated microbial systems. We discuss the strengths and limitations of various methods in analyzing microbiome composition, structure, and functionality, highlighting the potential of metagenomic data mining for practical applications. Finally, we address current limitations and challenges in microbiome study, and we discuss future trends toward the standardization and streamlining of microbiome study methodologies to accelerate progress in understanding microbiome functions and applications.
Key words: microbiome; metagenome; amplicon; analytical methods; functional exploration
微生物(细菌、真菌、古菌、病毒等)无处不在,对地球上各种生命形式的生存和发展具有不可或缺的影响,作为一个复杂多样的生态系统,微生物组在生态平衡、生物健康等方面发挥着关键作用。高通量测序技术出现之前,微生物研究主要依赖于分离培养技术,极大地限制了未培养微生物的研究。二、三代高通量测序技术的诞生打破了这一局限,开启了微生物研究的黄金发展时代,并催生出“微生物组(microbiome)”这一术语,其定义亦从特定环境中的微生物群落扩展至所有微生物及其基因信息的总和,为研究微生物的群落结构、功能特征及其与环境的关系提供了广泛视角。随着研究的深入,微生物组学在研究理念、基础实验和分析技术方面都取得了重要进展,二代高通量测序数据被细分为扩增子、培养组和宏基因组等数据,而三代高通量测序数据则进一步分为长扩增子、单菌基因组和三代宏基因组等数据。这些研究常常生成数百Gb(giga bases)乃至上Tb级的数据,在数据类型多样和数据量快速增长的背景下,如何从成千上万的宏基因组读序中提取出有效信息,揭示微生物的群落构成、功能作用和演变规律,是当今微生物组研究亟需解决的问题。在这种背景下,本文将系统梳理二代和三代高通量测序技术在微生物组研究中的应用方法,指出现有方法面临的挑战及潜在改进方向,旨在为微生物组数据处理的标准化和流程化提供参考,并以实例探讨如何更有效地利用微生物组数据解决实际应用中的问题。
1 微生物组研究进展
微生物(细菌、古菌、病毒、真菌等)无处不在,是地球上最具丰富性和多样性的有机体,对人类和动植物健康、生态环境、工业制造和农业生产等领域至关重要(图1-A)。微生物组一术语最初指在特定栖息地中具有独特理化特性的微生物群落[1-2],随后这一概念被扩展为特定环境或生态系统中所有微生物及其遗传信息的总和[3]。在过去的十几年中,微生物组的研究取得了前所未有的进展,二、三代高通量测序技术和分析方法的进步不断革新了微生物领域的重大发现,掀起了国际学术界基于宏基因组方法研究微生物组的热潮。文献检索结果显示,2006年迄今测序方法和微生物组文章呈井喷式增长趋势(图1-B),其中人类微生物组研究最多(使用以下关键词构建检索式,在Web of Science核心库中进行文献检索:“‘Amplicon’ or ‘16S rRNA’ or ‘16S rDNA’ or ‘ITS’ and ‘sequence’ and ‘microbiome’”; “‘metagenome’ and ‘second generation’ or ‘shotgun’ or ‘high-throughput’ and ‘microbiome’”; “‘Pacbio’ or ‘Nanopore’ or ‘long-read’ or ‘the third generation sequence’ and ‘microbiome’”)。
然而,随着多样的微生物组研究算法和分析工具的发展(图1-C),如何合理采用最优手段,挖掘海量数据的有效信息,解决科学和实际问题,是目前本领域研究面临的重要挑战之一[4]。本文通过系统梳理二、三代高通量测序研究的主要分析方法,通过解析其在“植物与根系微生物的互作机制”等应用流程,旨在提升微生物组方法的标准化和流程化,提出挖掘样本中微生物组成、结构和功能的应用视角。
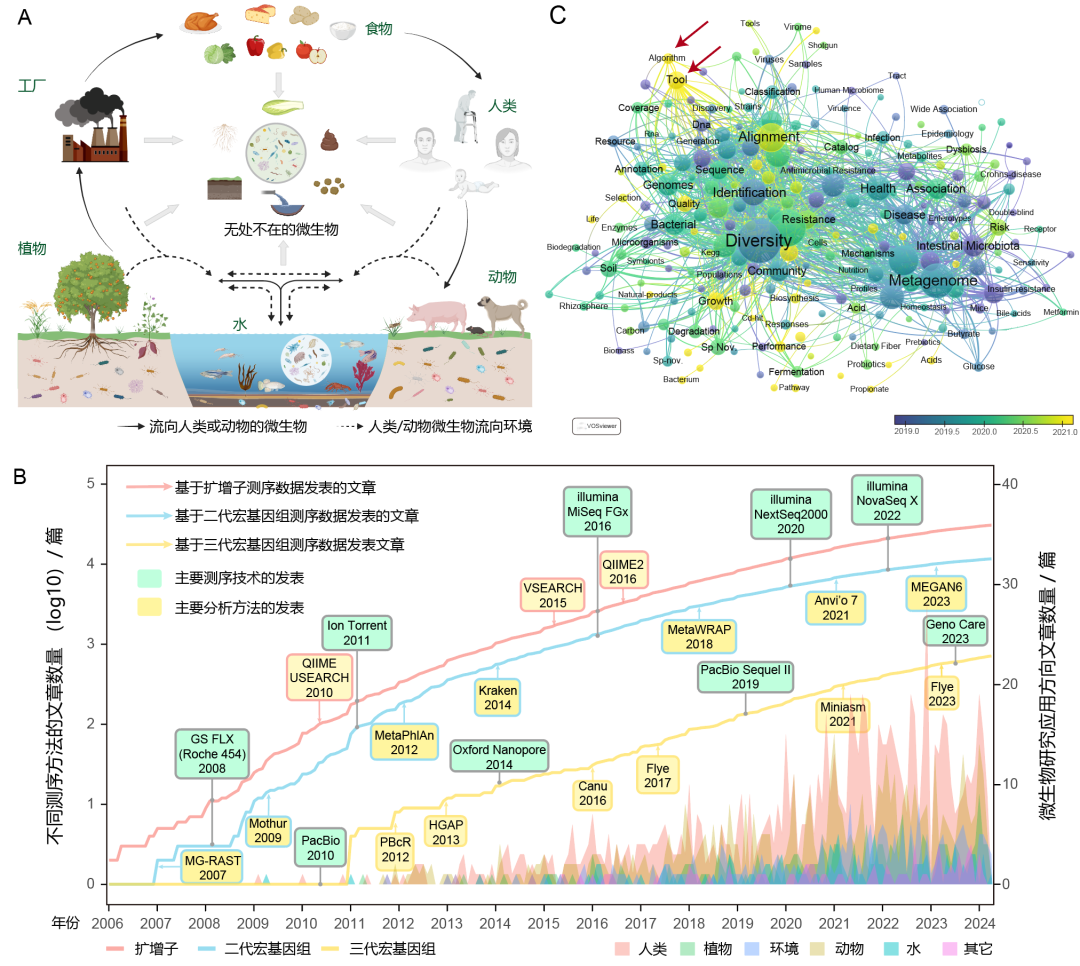
A:微生物组研究的应用场景;B:重大的技术和方法发展推动微生物组的研究;C:微生物组研究热点的发展趋势
A: Application scenarios of microbiome study. B: Significant technological and methodological developments driving microbiome study. C: Trends in key areas of microbiome study
图1 微生物组研究的发展与应用
Fig. 1 Development and application of microbiome study
2 微生物组二代高通量测序分析方法和应用
二代测序(next generation sequencing, NGS),又被称为高通量测序,采用合成测序(sequencing by synthesis)原理,主要步骤为基因组片段化、随机扩增建库和测序,这种技术具有特异性高、通量大、成本低等优点。常见的二代测序平台为华大智造、illumina、Ion Torrent、真迈生物等,其中illumina和华大占据主流(图1-B)。由于不受微生物培养和筛选过程的限制,加之高通量测序技术的迅猛发展,二代高通量测序已被广泛地应用于研究人类、动植物、环境等样品微生物组的群落变化。扩增子、培养组和宏基因组等数据分析是二代高通量测序的常见手段,其中扩增子分析通过靶向特定的微生物基因序列,来评估微生物的多样性;培养组分析为结合多种培养条件和二代高通量测序(多为扩增子测序),用于微生物培养和鉴定;宏基因组分析则以特定环境中所有微生物的基因组[5]为研究对象,可深入获取微生物物种或株系的基因结构,比扩增子分析更全面地揭示微生物的物种和功能组成。
2.1 扩增子分析
高通量扩增子分析是针对微生物组中特定基因组区域(如16S rDNA、18S rDNA、ITS等基因片段)中的基因变异进行分析,以揭示微生物群落的物种组成、多样性和分布规律等特征[6],其已成为研究低丰度变异或高异质性遗传学的普遍方法[7]。首先,从提取的DNA经PCR扩增获取靶向序列,进一步通过建库测序获取原始数据(raw data)。随后经过质控、去接头、拼接和去嵌合体等操作后,获取纯净数据(clean data),然后采用USEARCH[8]或DADA2[9]分别获取OTUs(operational taxonomic units)或ASVs(amplicon sequence variants)特征表,最后根据数据类型对特征表进行抽平处理,用于后续的下游分析(图2),如稀释曲线分析、物种组成及丰富度、多样性分析、物种差异比较、功能预测等内容。
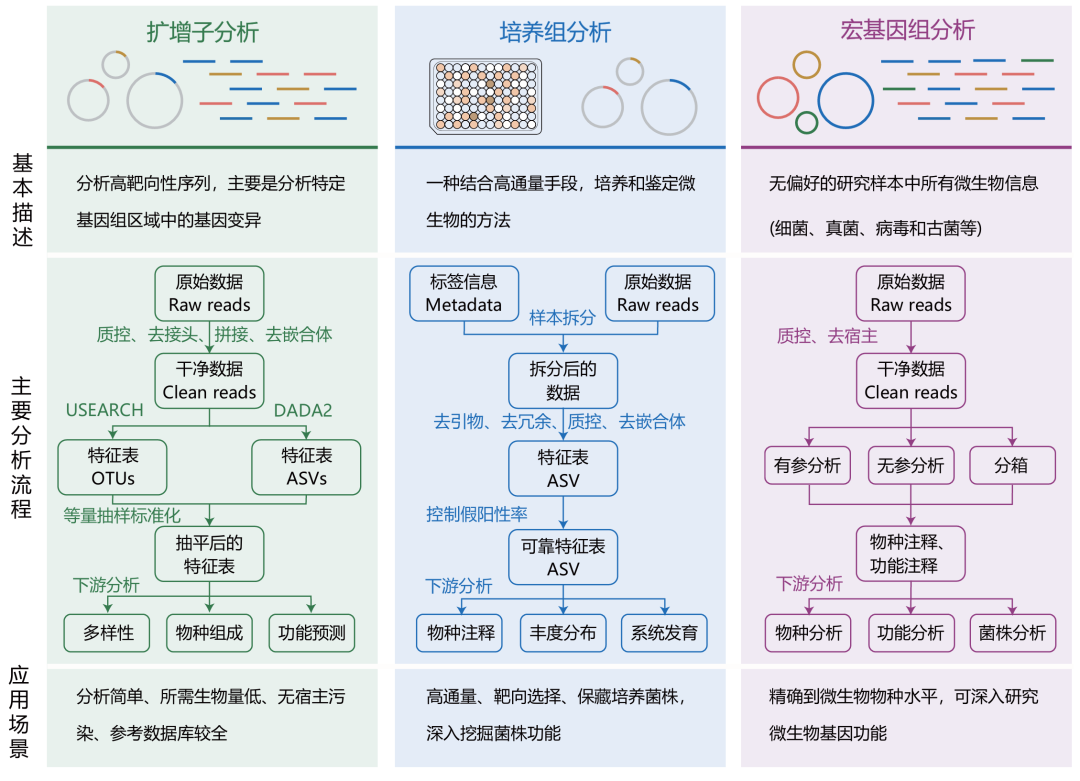
图2 基于二代高通量测序技术的分析流程和应用方案
Fig. 2 Analysis workflow and applications based on second-generation high-throughput sequencing technology
扩增子测序结果的数据量小,参考数据库覆盖较全面,分析技术相对较成熟[10]。此外,扩增子分析还具有所需的生物样本量少、测序成本低、宿主污染影响小等优点,已在人类、动植物、环境微生物群落检测等领域得到广泛应用(图1-B)。目前,已有QIIME[11]、QIIME 2[12]、MOTHUR[13]、DADA2[9]、EasyAmplicon[14]等多种成熟的软件或流程用于扩增子数据分析,推动各个领域对复杂的微生物群落组成及动态变化的深入认知。然而,受限于靶向片段的长度,扩增子的数据分辨率有限,许多序列只能注释到属级别的分类水平[6, 12, 15],因此扩增子分析的主要应用仍集中在微生物物种组成、进化关系和群落多样性变化等方面。
2.2 培养组分析
绝大多数的微生物都是难培养或未培养微生物,使得人类对微生物的认知不足1%,高通量测序为快速培养和鉴定微生物提供了绝佳机遇[16]。培养组的简要步骤为对含微生物的溶液进行稀释、培养和保存,采用碱裂解法提取96孔板中培养的细菌基因组,以及两步扩增法对每个样本的特异性引物加入标签和接头,随后对纯化混合后的样本开展二代高通量测序[17]。测序数据通过双端合并、标签拆分、引物切除、挑选代表性序列、去冗余、质控、去除嵌合体后,进入物种注释、进化关系等下游分析阶段(图2),以实现对大量微生物的快速鉴定。培养组完整流程中,虽然采用了扩增子技术,但与常规扩增子分析相比,其重点在于微生物的鉴定,且其数据分析过程涉及标签拆分,因此具有独特的数据处理流程[17]。
目前,已经针对细菌16S rDNA发表了详细的培养组实验和分析流程[17],对于其他类群或片段的培养组分析,只需替换代码中微生物标签信息即可。此外,MutiplePrimer网站[18]的开发,为用户设计特异性引物提供了便捷方案。培养组的发展不仅有助于我们快速鉴定培养菌株,并深入挖掘其功能,还能通过预测菌株的代谢特性,发现促进或抑制菌株生长的添加物,从而获得难培养的微生物[19]。未来,开展合成菌群研究或将成为培养组研究的发展趋势。对于感兴趣的菌株,我们可以进行基因组测序、基因敲除验证等实验,以进一步揭示其生理、形态和功能特征。
2.3 宏基因组分析
宏基因组是以特定环境中的整个微生物群落(细菌、真菌、病毒和古菌等)为研究对象,无需分离培养,直接从环境样本中提取DNA进行建库和测序,从而避免了微生物组的可培养性、引物设计或扩增偏好性带来的问题,能更真实地反映微生物群落的信息[20]。原始数据通过质控、去宿主等操作后,获取纯净数据,随后可进行有参分析、无参分析(组装)和分箱等操作[15]。根据物种和功能注释结果,可以开展下游物种、功能和菌株等深入分析(图2),用于研究环境微生物的群落结构、物种分类、系统进化、基因功能和代谢网络等。
宏基因组分析可在扩增子分析的基础上,获取微生物宏基因组组装基因组(Metagenome-Assembled Genome, MAG),进而深入研究物种、基因和功能层面。然而,宏基因组分析严重受所选数据库的影响,如Kraken2[21]不同数据库之间存在差异,HUMAnN 2[22]或HuMAnN 3[23]的数据库中以细菌的类群为主等。此外,由于宏基因组测序是对提取到的DNA全部测序,所以对于高宿主的污染样本,会获取到大量非目标序列,这些序列的分析不仅影响了下游数据的准确性,还消耗了不必要的计算资源,为此,研究团队开发了HostPurge的流程(https://github.com/HaoLuo-leo/HostPurge),以期提供一种快速、准确的去宿主方案。
2.4 二代高通量测序分析植物与根系微生物组的互作
植物与微生物之间的互作关系密切又复杂,研究这一现象和机制是生命科学研究的热点问题,也是发展农业微生物产业新生态的关键。目前中国科学家白洋及其团队等在该领域已取得突破性进展[24],文章利用微生物组二代高通量测序技术,涵盖扩增子、培养组和宏基因组的手段,探讨了微生物组影响水稻营养吸收的研究框架(图3),为同行挖掘植物与微生物组的互作关系提供了研究思路。
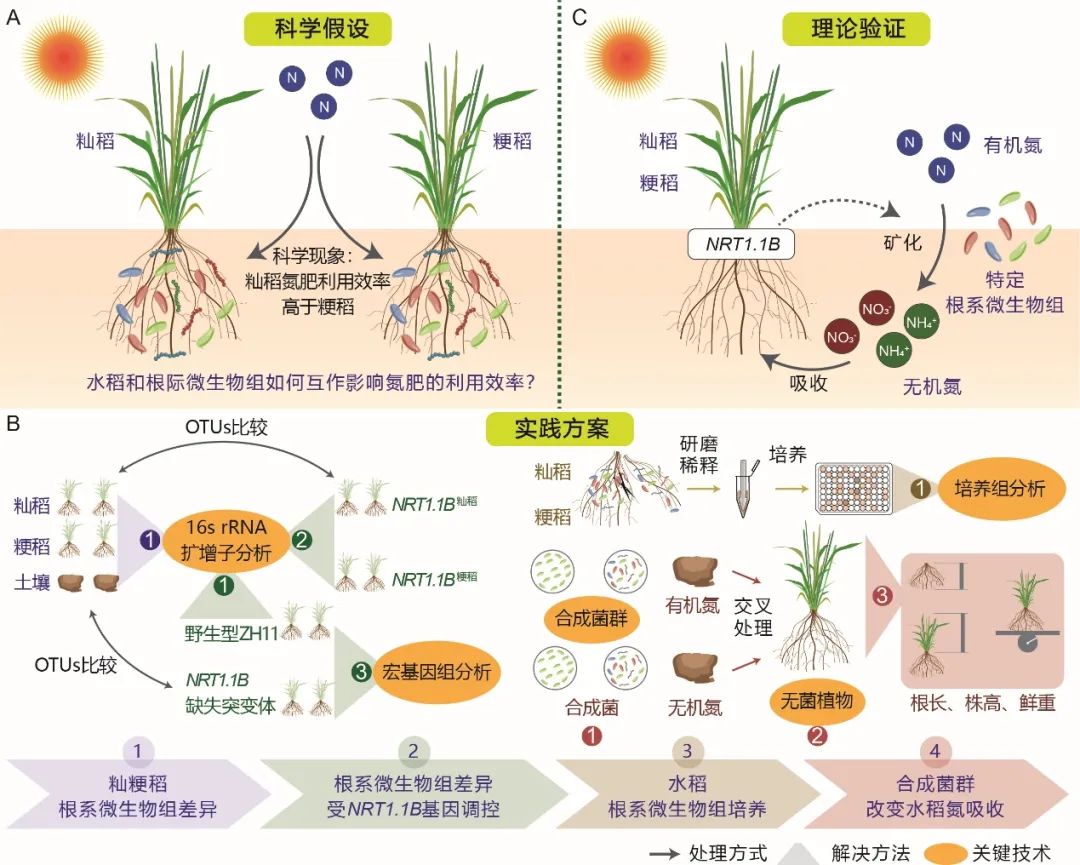
A:提出科学假设;根据籼稻氮肥利用效率高于粳稻这一科学现象提出,水稻和根际微生物组如何互作影响氮肥的利用效率这一科学假设;B:开展实践方案;基于扩增子、宏基因组、培养组分析,发现籼粳稻根系微生物组差异分析、根系微生物组差异受NRT1.1B基因调控、水稻根系微生物培养和合成菌群改变水稻氮吸收实验的实践方案;C:获取理论验证;特定根系微生物会参与有机氮矿化的过程促进水稻的氮元素吸收,且该现象与硝酸盐转运蛋白基因NRT1.1B在籼粳稻之间的自然变异相关联
A: Develop scientific hypotheses. Based on the observation that nitrogen use efficiency of indica rice is higher than that of japonica rice, the hypothesis that rice and the rhizosphere microbiome interact to influence nitrogen use efficiency is proposed. B: Implement practical programs. Utilizing amplification, metagenomic analysis, and culture group analysis, the differences in root microbiomes between indica and japonica rice are examined. Additionally, the role of the NRT1.1B gene in regulating these differences is investigated, and experiments are conducted on the cultivation of rice root microbiomes and synthetic communities to assess changes in nitrogen absorption. C: Theoretical validation. Specific root microbiomes participate in the process of organic nitrogen mineralization, thereby enhancing nitrogen uptake in rice. This phenomenon is linked to the natural variation of the nitrate transporter gene NRT1.1B between indica and japonica rice
图3 基于宏基因组分析探讨植物与根际微生物的互作对植物营养吸收影响的案例
Fig. 3 Case study on the effects of plants-rhizosphere microorganism interaction on plant nutrient uptake based on metagenomic analysis
亚洲栽培稻(Oryza sativa)主要分为籼稻(indica)和粳稻(japonica),多项实验表明籼稻通常比粳稻具有更高的氮利用效率。根据这一科学现象,作者提出水稻和根际微生物组的互作,影响了水稻从土壤中吸收氮的效率这一科学假设,并开展了多项实验验证,验证其理论假设。包括:(1)基于扩增子,分析了不同稻亚种根际微生物差异,利用随机森林(random-forest)方法筛选出特定微生物组作为生物标志物,并发现籼稻富集的OTUs与氮循环相关。(2)比较NRT1.1B籼稻和NRT1.1B粳稻、野生型ZH11和NRT1.1B缺失突变体根际微生物的扩增子数据,发现籼稻富集的OTUs与NRT1.1B基因调控的微生物重合,并结合宏基因组数据分析发现微生物功能在氮循环相关的通路上存在差异。(3)基于培养组技术,建立了籼稻和粳稻的可培养根系微生物组菌库。(4)通过根长、株高和水稻鲜重等植物生长指标,比较了不同的合成菌群、氮素类型对无菌稻种氮吸收效率的影响,验证了特定根系微生物组参与有机氮矿化,促进水稻的氮元素吸收的科学假设。通过充分利用扩增子分析、宏基因组、培养组等高通量测序技术,研究系统筛选和鉴定了对氮循环有重要作用的微生物,并创建了功能性微生物菌库,为调控微生物组以提高作物养分利用效率提供新的方法和思路。
3 微生物组三代高通量测序分析方法和应用
尽管二代高通量测序已广泛应用于各个领域(图1),但其读序短、拼接难是不可回避的技术难点[6]。随着测序技术的发展,三代测序技术应运而生,并以PacBio公司的单分子实时测序技术(SMRT)和牛津纳米孔公司(ONT)的纳米孔测序技术为主流,为解析宏基因组数据中复杂基因区域、组装完整的高质量基因组提供了全新技术。目前三代高通量测序被广泛应用于长扩增子测序[25]、单菌基因组测序[26]以及微生物宏基因组测序等领域,为微生物组学和生命科学领域带来了新的突破。
3.1 长扩增子分析
基于二代扩增子测序,最多可以获取500 bp的可变区域片段[7],然而常用于识别的细菌16S rDNA片段,其全长在1 500–1 600 bp[27],因此二代扩增子测序通常只能覆盖16S rDNA可变区域的部分片段,如V1–V3、V4或V5‒V7,限制了物种检测的分辨率。尽管一代Sanger测序可以获取完整的16S基因片段,但面对大量样本时,则显现出成本高、效率慢的弊端。三代扩增子技术的开发利用了三代长读序的优势,使其可检测特定长片段基因区域的基因变异[25],显著提高了从生物量低、复杂度高的微生物组样本中,对微生物物种或菌株水平上鉴定。
目前,PacBio环形CCS技术(circular consensus sequencing)可以对长片段进行测序和错误校正,提高了微生物组数据在物种级别上的高保真度。其主要分析流程为,使用SMRT-Link分析软件对下机数据进行拆分(demultiplex)和质控,过滤掉低质量或非目标序列,并删除小于1 300 bp和大于1 700 bp的序列[6]。然后,采用DADA2[9]去除引物、嵌合体后,进行分类注释获取特征表,开展下游分析[28]。同时,也有多个团队基于牛津Nanopore和国产齐碳纳米孔技术,开展了长扩增子测序,获取的原始文件经过碱基识别后转为fastq数据,并经质控、分类注释后,开展物种组成、多样性和网络分析等(图4)。由于长扩增子测序在分类鉴定和群落多样性研究中的优势,被广泛应用于临床诊断[29]、病原菌快速检测[30-31]、微生物类群鉴定[32]等。
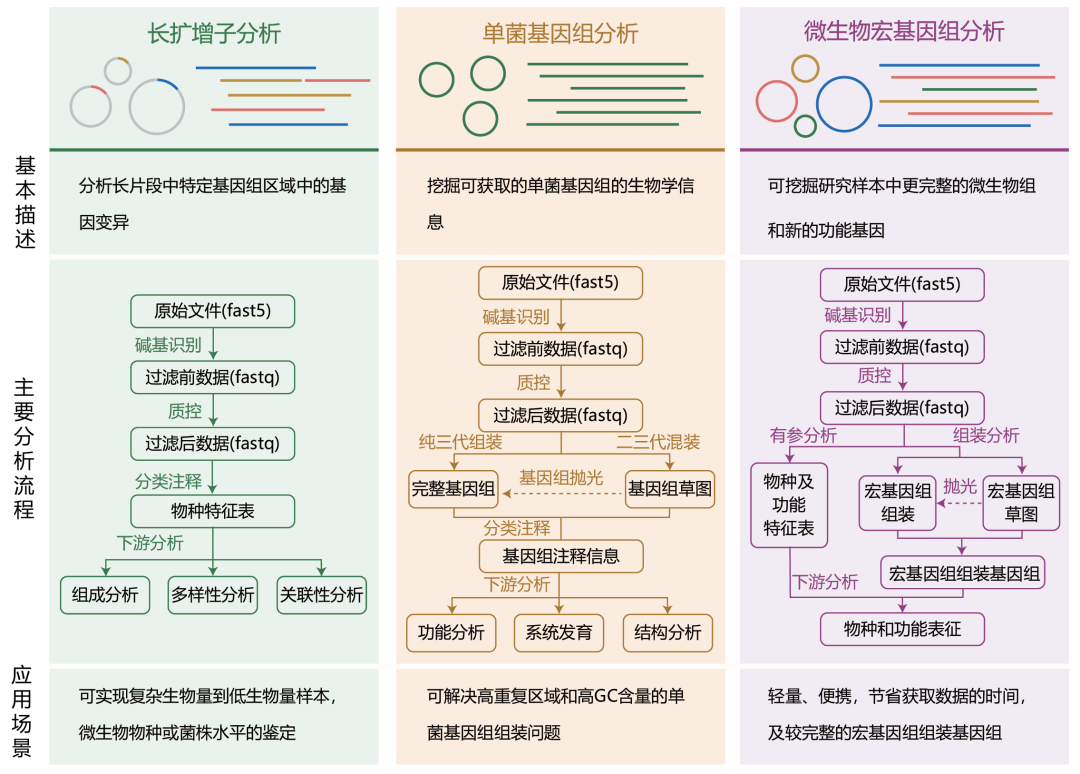
图4 基于三代纳米孔技术的分析流程和应用方案
Fig. 4 Analysis workflow and applications based on third-generation nanopore technology
3.2 单菌基因组分析
目前仅依靠二代宏基因组测序,很难解决单菌基因组中的高重复区域和高GC区域的组装问题,因此组装后的单菌基因组往往不完整。目前纳米孔测序技术已逐渐成为组装单菌基因组的常见手段,然而尽管三代测序具有用时短、读序长的优势,但是其碱基识别准确度仍存在较高错误率(<15%)[33],且成本相对较高,限制了其现阶段的大规模应用。二、三代测序数据的混合组装、互取所“长”,既可保证所获基因组的完整性,又可实现基因组碱基的准确性,也已成为学术界的常见选择[26]。为挖掘单菌基因组的生物学信息,过滤后的数据经过纯三代组装或二、三代混合组装后,开展分类注释,并对单菌基因组的功能、结构和系统发育等进行下游分析(图4)。
随着三代测序技术通量的不断提升,牛津纳米孔的MinION测序通量从最初的数百兆增加到10–15 Gb,而后续PromethION的发展则可实现150 Gb的通量[34],使其在单菌基因组中的应用从细菌等小型基因组,逐渐扩展到真菌等大型基因组。同时,纳米孔的测序精度也从87%提升到98.3%[35],大幅增强了纳米孔测序的准确度。此外,经二代数据校正后,三代数据基因组草图(draft genome)准确率可达99.9%[36],这些技术的发展必将进一步促进单菌基因组在物种识别、基因分析、菌株相关性和病原菌传播模式研究中的应用。
3.3 三代宏基因组分析
与二代宏基因组测序相比,三代宏基因组技术能够获取几乎完整的宏基因组组装基因组(MAG),从而有助于发现更多未知微生物和新的功能基因[37, 38]。同时,纳米孔测序平台轻量、便携的优点,显著缩短了从样本采集到宏基因组信息获取的时间,这对流行病的暴发预警以及病原体的快速检测尤为重要。微生物三代宏基因组分析流程相对简单,包括质控、过滤、去宿主,然后开展有参分析和组装分析。目前,三代宏基因组数据的有参分析软件多参考了二代,如使用Kraken2[21]进行物种注释。三代宏基因组组装则类似于三代单菌组装,可进行纯三代组装或二、三代混合组装,然后进行下游的物种和功能表征。针对三代宏基因组数据的复杂性和多样性,开发了多种适用于长读序的生物信息软件,如flye[39]、canu[40]、wtdbg2[41]、NextDenovo[42]等纯三代组装软件,以及OPERA-MS[43]、metaSPAdes[44]、unicycler[45]等二、三代混合组装软件,扬州大学联合中国农科院基因组所也针对三代宏基因组数据开发了相关分析流程EasyNanoMeta(https://github.com/P-kai/EasyNanoMeta),以推动长读序数据的高效利用。
尽管针对长读序数据已开发了部分生物信息学软件和方法,如错误矫正方法[46]或混合组装矫正方法[47],但三代宏基因组分析仍处于发展初期,基于短读序算法的三代宏基因组分析,在默认参数下很难适用于长读序数据集[37],可能导致我们解读纳米孔测序的读序时存在较高的错误率。因此,未来亟需建立适合三代宏基因组数据分析的“金标准”。
3.4 三代高通量测序揭示微生物组的多样性
长读测序弥补了短读测序在处理重复区域、高度同源区域等方面的不足,实现了更为完整的基因组分辨率[37-38]。在微生物组数据中,常常存在多种亲缘关系接近的菌株,其基因组仅在少数几个位置存在差异,导致样本中的分类变化十分复杂[48-49]。除非同时对其相邻区域进行测序,很难通过计算处理这些基因组间的相似或重复片段,长读测序成为克服这些问题的一种潜在策略,从而提高读序覆盖生物特定区域的可能,并被广泛应用于获取完整的MAG,以及功能基因(如抗性基因[50])的检测。此外,因三代测序技术轻便、便携的特点,可显著节省数据获取的时间,在病原体识别领域展现了其应用潜力[48, 51]。然而,相较于二代测序,三代测序通常对DNA的质量要求更高[48]、测序通量小、碱基识别准确度低,因此,二、三代混合测序分析仍是当前的主要选择。
4 展望
微生物组研究面临前所未有的机遇期和窗口期,随着人工智能、超分辨率光学成像、多组学等前沿技术的成熟,多学科交叉融合正在打破技术壁垒,进而驱动微生物组的深度发展,然而,微生物组研究仍旧面临多方面的限制和挑战。首先,现有研究主要集中于细菌群落,忽视了对真菌、病毒、原生动物等“暗物质”微生物的探索,这些微生物在环境中的广泛分布和寄生现象表明其可能具有与细菌同等的重要生态功能,因此未来亟需加大对微生物“暗物质”的研究 [52]。其次,全球微生物组数据库和高质量的宏基因组组装菌种库建设尚不完善,限制了对微生物多样性和跨区域传播的解析。未来应进一步加快菌种库的建设,推动数据共享,构建具有全球代表性的微生物组数据库,以支持微生物生态学的深入研究。
目前,宿主基因污染仍是显著影响数据准确性的因素之一,在数据处理方面,现有的过滤算法(映射比对或k-mer)尚未彻底消除这一干扰,亟需引入新的过滤算法以提高分析的可靠性。此外,不同数据分析流程的差异性,限制了研究间的稳定性、可重复性和结果的可比性,且当前研究多聚焦群落组成,而忽略了基因通路、蛋白质功能和代谢网络的系统性分析,这些信息对理解微生物生态功能和致病机制至关重要。尽管已有诸多工具和自动化流程(如EasyAmplicon、EasyMetagenome和Wekemo Bioincloud)支持二代数据的标准化[4, 14-15],但随着新一代测序技术的发展,未来的数据分析流程仍需在“暗物质”挖掘、功能分析、多组学互作和下游可视化等方面进一步优化,建立统一数据分析流程的“金标准”,以提升分析准确性和可重复性,揭示微生物群落在生态系统中的功能网络及其作用机制。
原文链接:https://biotech.aiijournal.com/CN/10.13560/j.cnki.biotech.bull.1985.2024-0788
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文

















 1760
1760

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









