







研究论文
● 期刊:Cell (IF:45.5)
● DOI: https://doi.org/10.1016/j.cell.2024.12.034
●原文链接: https://www.cell.com/cell/fulltext/S0092-8674(24)01477-6
● 第一作者:Fuyong Li(李福勇),Anissa M. Armet
● 通讯作者:Jens Walter(jenswalter@ucc.ie)
● 发表日期:2025-1-23
● 主要单位:
加拿大阿尔伯塔大学农业、食品与营养科学系、浙江大学动物科学学院
摘要Abstract
工业化对肠道微生物群产生不利影响,使个体易患慢性非传染性疾病。我们在加拿大健康成年人的随机对照喂养试验中测试了一种微生物群恢复策略,包括一种重现非工业化饮食模式关键特征的饮食(恢复饮食)和一种在工业化微生物群中很少发现的细菌-罗伊氏乳杆菌(Limosilactobacillus reuteri)。尽管恢复饮食降低了肠道微生物群的多样性,但它增强了源自巴布亚新几内亚农村地区的罗伊氏乳杆菌菌株(PB-W1)的持久性,并纠正了受工业化影响的几种微生物组特征。这种饮食还有益地改变了与慢性非传染性疾病相关的微生物群衍生血浆代谢物。除了提高罗伊氏乳杆菌的定植之外,恢复性饮食还带来了显著的心血管代谢益处,其中许多益处可以通过基线和饮食响应性微生物组特征准确预测。研究结果表明,针对恢复肠道微生物组的饮食干预可以改善宿主与微生物组之间的相互作用,这种干预可以缓解可能由互作引起的慢性疾病的发生。这一发现可以指导饮食建议以及治疗和营养策略的制定。
结果Results
微生物组恢复策略的设计
本研究之所以选择罗伊氏乳杆菌(L. reuteri)进行研究,是因为它在工业化社会的人类消化道微生物组中较为罕见;其在巴布亚新几内亚农村居民的粪便微生物组中占优势地位,但在美国对照组的受试者中却未被检测到。该细菌被普遍认为是安全的,并具有明确的健康促进作用。为了验证“源自非工业化人群的罗伊氏乳杆菌菌株会对非工业化类型的饮食表现出更高的适应性”这一假设,我们比较了源自巴布亚新几内亚农村的罗伊氏乳杆菌菌株(“PB-W1”)与源自德国的罗伊氏乳杆菌模式菌株(“DSM 20016T”)。这两个菌株属于罗伊氏乳杆菌的不同系统发育谱系和亚种,其平均核苷酸一致性(ANI)值仅为96.2%,因此存在遗传差异。
我们所设计的这种恢复饮食基于巴布亚新几内亚农村居民的常见饮食,这些食物同时在加拿大也可获得(如豆类、红薯、大米、黄瓜和卷心菜),以及含有高含量棉子糖和水苏糖的食物(如菊芋、豌豆和洋葱),这些成分是罗伊氏乳杆菌的生长底物。该饮食具有与非工业化饮食模式相似的关键特征,在一定程度上模拟了我们祖先的饮食结构特点:以植物为主,不含乳制品和小麦,限制精加工食品的摄入,低血糖指数和能量密度,以及超过推荐量的膳食纤维(每1000千卡含22克)。恢复饮食所提供的总能量中,约60%来自碳水化合物,15%来自蛋白质,25%来自脂肪,这些比例均在可接受的宏量营养素分布范围内。
一项旨在表征肠道微生物组恢复策略效果的人体试验
我们对健康成人(n = 30)进行了一项随机对照喂养试验,以测试我们的微生物组恢复策略对肠道微生物组和宿主代谢的影响(图1A)。我们的目标是同时确定:(1)罗伊氏乳杆菌是否可以在西方人群的肠道微生物组中重新建立及其对宿主的影响 (2)不含罗伊氏乳杆菌补充的恢复饮食自身对于人体的作用 (3)罗伊氏乳杆菌和恢复饮食之间潜在的有益相互作用。受试者被随机分配,其中一组接受为期3周的恢复饮食——这是能够满足个人热量需求的标准化膳食,4天一轮换(表S1;示例如图S1所示);另一组别在这3周中保持其常规饮食和摄入量,通过重复24小时饮食回忆进行监测(表S2)。经过上述3周的试验后,两组经过不受饮食干预的3周“洗脱期”。此后,两组受试者的饮食互换,具体干预方法同前述,为期3周。然后进行最后的3周洗脱期。在平行组设计中,受试者在每个饮食期的第四天(试验的第4天和第46天)随机接受1010个L. reuteri PB-W1或DSM 20016T活细胞或安慰剂(麦芽糊精)的单次接种。
从2019年1月到2020年1月,我们共筛查了266人,对42人进行了资格评估(有关纳入和排除标准,请参阅STAR方法)。在评估的42人中,只有1人的粪便中检测到罗伊氏乳杆菌,证实了罗伊氏乳杆菌在西方人群的肠道中很少见。41名未检测到罗伊氏乳杆菌的个体被随机分组,其中30名受试者(年龄28.2 ± 6.7岁,体重指数[BMI]23.8 ± 2.6kg/m2[平均值 ± SD];基线特征见表S3)接受并完成了上述饮食干预,被纳入后续统计分析,其余的11名受试者因个人原因(n = 7)、依从性低(n = 2)或不愿忍受胃肠道症状(n = 2)退出(图1B)。平均而言,与受试者的常规饮食相比,恢复饮食期间的纤维摄入量增加了一倍(图1C),而饱和脂肪则显著减少(表S2),但能量摄入量没有显著变化(图1D)。
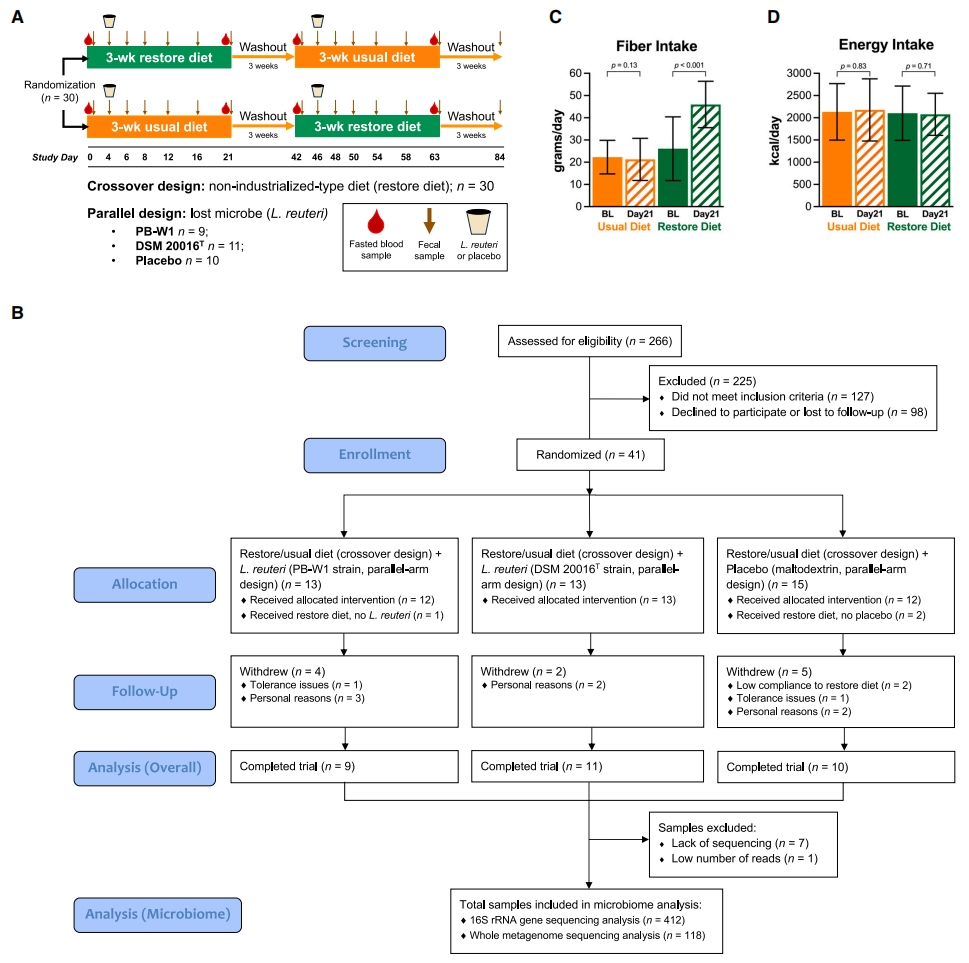
图 1 | 健康成年人微生物群恢复策略的实验测试
(A)随机对照试验设计和样本类型收集的描述。
(B)研究流程的CONSORT图。
(C和D)每个饮食期间(C)纤维和(D)能量摄入量的变化以条形图显 示,平均值 ± SD;配对t检验,p < 0.05。另见图S1和表S1-S4。BL,每个饮食期的基线;Day21,每个饮食期的第21天;DSM 20016T,L. reuteri模式菌株;PB-W1,来自巴布亚新几内亚农村的L. reuteri菌株。
罗伊氏乳杆菌菌株的持久性和存活率
在摄入罗伊氏乳杆菌PB-W1或DSM 20016T补剂两天后,通过培养和定量PCR(qPCR)可从受试者的粪便中检测到罗伊氏乳杆菌,但在安慰剂组中则未检测到(图2A和S3A)。在两个饮食期中,在摄入PB-W1两天后,粪便中细菌数量(基于培养和qPCR,分别为7.64 ± 0.11和7.85 ± 0.15 Log10细胞/g)均高于DSM 20016T(基于培养和qPCR,分别为6.36 ± 0.11和6.66 ± 0.20 Log10细胞/g)(培养和qPCR的p < 0.001,配对t检验)(图2A和S3A)。随着时间的推移,粪便中两种罗伊氏乳杆菌菌株的丰度均下降。在给药后12-17天内,多数受试者的粪便样品中罗伊氏乳杆菌的丰度均已降至不可检测水平,除了一名受试者。尽管丰度较低,PB-W1在整个试验过程中都稳定地定植于该受试者体内(图S3B)。恢复饮食提高了样本中PB-W1的丰度,但没有提高DSM 20016T的丰度,在给药后4天(6.61 ± 0.24对比5.78 ± 0.37,基于培养,p = 0.02)和8天(3.90 ± 0.35对比2.40 ± 0.24,基于培养,p = 0.02),并且PB-W1细胞数量在这些时间点均显著高于DSM 20016T(基于培养,两天p < 0.001;基于qPCR,给药后4天和8天p = 0.003和0.02)(图2A和S3A)。鉴于饮食对PB-W1的可检出率在培养物(活细菌)中比在qPCR数据(活细菌和死细菌)中更强,我们猜想恢复饮食提高了PB-W1的存活率。事实上,在恢复饮食条件下,PB-W1菌株的培养与qPCR计数比值明显更高(分别为给药后2天和4天p = 0.02和0.05;配对t检验),但对于DSM 20016T却非如此(图2B)。总而言之,这些结果表明,PB-W1相较于DSM20016T有更高的生态适应性和存活率,尤其是在给药后的早期阶段,且这种优势能够被恢复饮食进一步放大。然而,由于两种菌株在肠道中的持久性均较低,这种相对优势也是短暂的,仅在一个样本中出现了PB-W1的长期定植。
我们将罗伊氏乳杆菌接种物作为肠道微生物组下游分析中的固定因素,使用线性混合模型或双因素重复测量方差分析,但这些分析并未检测到罗伊氏乳杆菌接种物对肠道微生物组组成、多样性或功能特征的任何显著影响(未显示数据)。
恢复饮食对肠道微生物群落的影响
经过对16S rRNA基因扩增子测序(基于扩增子序列变体[ASV])和整个宏基因组测序(WMS,基于物种级基因组箱[SGB])的评估,我们认为恢复饮食降低了肠道微生物群的alpha多样性:以香农指数(基于ASV,p < 0.001,线性混合模型;基于SGB,p = 0.003,双因素重复测量方差分析)(图2C和S3C),观察到的特征数量(ASV和SGB的p < 0.001)(图2D和S3D),以及Pielou均匀度(基于ASV,p < 0.001;基于SGB,p = 0.024)(图S3E)为评估指标。α多样性的减少发生在恢复饮食的前4天内,并在整个饮食期间保持稳定(图2C、2D和S3E)。此外,WMS预测功能特征也揭示了样本中α多样性的减少,例如观察到的通路数量减少(p = 0.013,双因素重复测量方差分析),碳水化合物活性酶(CAZymes)减少(p < 0.001)(图2D),而这些功能特征的Shannon指数和Pielou均匀度没有发生显著变化(图S3C和S3E)。饮食干预并未改变功能酶(ECs)的α多样性指数(图S3C-S3E)。
通过比较治疗时间点与基线之间的β多样性(Bray-Curtis距离)差异,结果表明恢复饮食显著改变了整个肠道微生物群落,这在ASVs(p < 0.001,线性混合模型;图2E)和SGBs(p < 0.001,双因素重复测量方差分析;图2F)中均可检测到。饮食干预诱导的基线与恢复饮食期第4天样本之间的β多样性差异同基线丰富度(观察到的ASVs数量;rs = –0.38,p = 0.04,Spearman等级相关)呈负相关。这表明,宿主肠道中原始的微生物群越多样(基线丰富度越高),肠道菌群对饮食变化的适应性越强(β多样性的前后改变越小),这佐证了多样性-稳定性假说以及先前研究中评估肠道微生物群对饮食干预反应的研究成果。16S rRNA基因扩增子测序的动态数据显示,微生物群落变化发生在恢复饮食干预介入的前4天内,并在整个恢复饮食过程中保持稳定(图2E)。恢复饮食引起的更高β多样性也被发现具有功能特征,能对代谢通路(p = 0.011,双因素重复测量方差分析)和碳水化合物活性酶(CAZymes) ([p = 0.040],ECs [p = 0.030])产生影响(图2G-2I)。通过评估个体之间的Bray-Curtis距离,我们发现恢复饮食进一步扩大了不同受试者间肠道微生物组成(ASVs和SGBs均为p < 0.001;Wilcoxon符号秩检验)和一些代谢功能方面(对于代谢通路p = 0.014,对于ECs p <0.001)的差异,但不包括CAZymes(图S3F)。
为了确定饮食在塑造肠道菌群方面与其他相关因素(例如个体差异和样本运输时间)相比的相对贡献,我们构建了一个Bray-Curtis距离矩阵,并通过主坐标分析(PCoA)将其投影到二维空间(图2J)。置换多元方差分析(PERMANOVA)显示,恢复饮食显著改变了整体微生物群落组成(p = 0.001,R2 = 0.015),但微生物群仍与个体表现出高相关性(p = 0.001,R2 = 0.774),这表明饮食仅解释了整个受试者群体中1.5%的微生物群变化(图2K)。因此,标准化饮食的干预并没有减少个体间的肠道微生物群差异,这与之前的研究一致。然而,饮食产生的影响至少是运输时间产生影响的5倍(0.2%的变化由排便频率解释,0.1%由粪便稠度解释;图2K)。
有趣的是,饮食变化解释了单个个体内22.6%–58.6%(p = 0.001–0.009)的微生物群变异(图2K)。这些发现表明,恢复饮食对肠道微生物群组成的调节作用受到个体差异的强烈影响。此外,“个体性”对微生物组功能特征的β多样性变异的贡献为62.8%–66.9%(p = 0.001)(图2L),而“饮食”的影响仅为1.7%–2.5%(p = 0.001;图2M)。这表明,尽管功能冗余部分掩盖了“个体性”在微生物组功能变异方面的重要作用(与直接分析肠道群落构成相比),但即使是从功能的角度来看,肠道微生物组仍然表现出显著的“个性特征”,与之相比饮食的影响甚小。
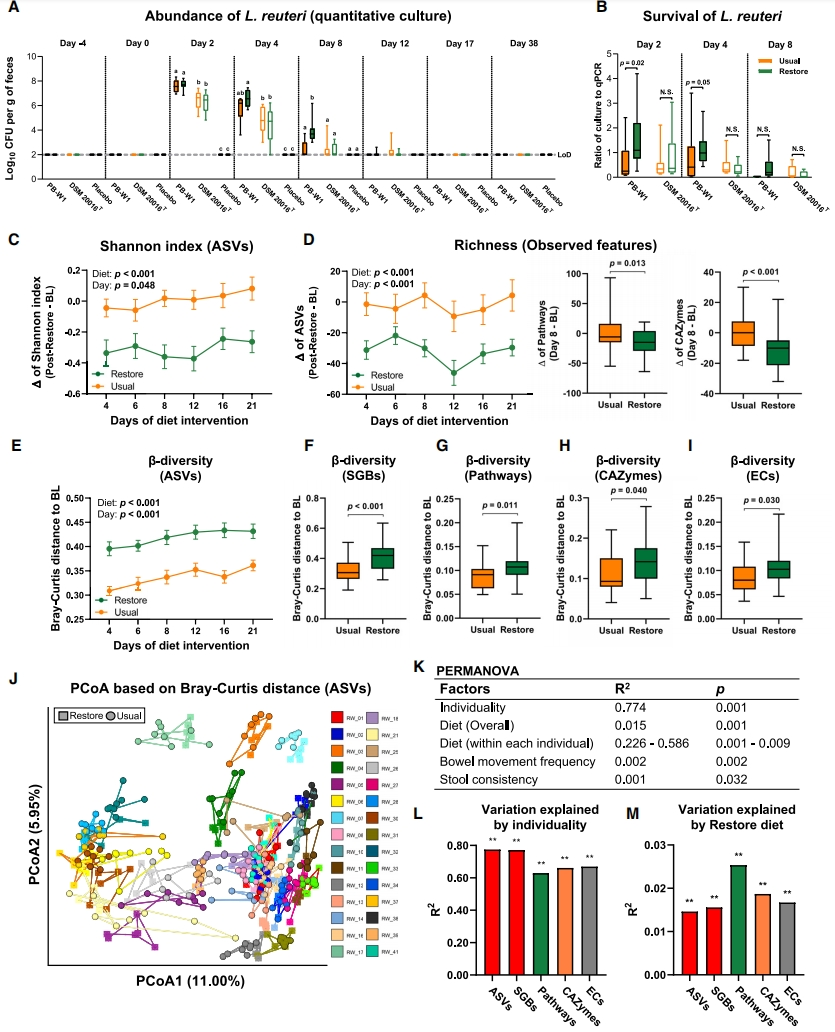
图 2 | 恢复饮食对肠杆菌持久性和微生物组多样性指数及变化的影响
(A)通过定量培养确定的粪便样本中罗伊氏乳杆菌的细胞数。数据以每克粪便中的细胞数Log10表示,在干预的每一天中,不同的字母表示基于双因素重复测量方差分析的显著差异,p < 0.05。
(B)罗伊氏乳杆菌的存活率,表示为在每个饮食期的第6、8和12天(即罗伊氏乳杆菌给药后2、4和8天)收集的粪便样本中使用定量培养估计的细胞数与qPCR的比率。选择此时间范围是因为到第16天(罗伊氏乳杆菌给药后12天),大多数样本中的罗伊氏乳杆菌都检测不到。在同一天内,分别应用配对t检验来比较恢复饮食和正常饮食期的罗伊氏乳杆菌PB-W1和DSM 20016T的存活状态;p < 0.05。
(C)根据ASV(线性混合模型)估计,基线和两个饮食期间每个受试者的微生物群之间的香农指数变化。
(D-I)(D)基于ASVs(线性混合模型)、通路(双因素重复测量方差分析)和CAZymes(双因素重复测量方差分析)估计的基线和每个饮食期间不同天数之间观察到的特征(丰富度)的变化。个体内(受试者内)Bray-Curtis距离(即每个饮食期间不同天数与受试者基线之间的距离),基于(E)ASVs(线性混合模型)、(F)SGBs、(G)通路、(H)CAZymes和(I)ECs(F-I的双因素重复测量方差分析)估计。
(J)使用基于ASVs的Bray-Curtis相异性矩阵对微生物群落进行PCoA分析(每种颜色代表不同的受试者)。
(K–M)(K)基于Bray-Curtis相异性矩阵的PERMANOVA进行了1,000次排列,以评估各种因素(即个体、饮食、排便频率和粪便稠度)对微生物群落结构的影响。微生物组成概况(ASVs和SGBs)和功能(通路、CAZymes和ECs)的变化由(L)个体和(M)恢复饮食解释;**p ≤ 0.001,PERMANOVA。数据呈现为箱线图或平均值 ± SEM。另见图S2和S3。ASVs,扩增子测序变体;BL,每个饮食期的基线;CAZymes,碳水化合物活性酶;DSM 20016T,罗伊氏乳杆菌型菌株;ECs,第4级酶分类;LoD,检测限;PB-W1,来自巴布亚新几内亚农村地区的罗伊氏乳杆菌菌株;N.S.,不显著;PCoA,主坐标分析;PERMANOVA,置换多元方差分析;SGBs,物种级基因组箱。病毒和细菌分类群的稀疏曲线。带状表示50次模拟中的最小和最大分类群。
恢复饮食对肠道微生物类群相对丰度及其编码功能的影响
在可检测到的ASVs中,有超过一半(122 / 205)在饮食干预期间发生了显著改变(FDR调整后p < 0.05,线性混合模型)(表S5)。对ASVs层次聚类表明,在恢复饮食期间收集的所有样本的平均值与在正常饮食期间收集的样本以及每个饮食期的基线分开聚类(图3A)。这表明,尽管上文检测到的个体差异很大(图2J和2K),但单个微生物类群的平均相对丰度仍然表现出显著的饮食诱导变化。
潜在的几种有益菌种(图3B)和属(图3C),包括双歧杆菌(Bifidobacterium)属(含三种双歧杆菌种(青春双歧杆菌(B. adolescentis)[ASV70和ASV187]、长双歧杆菌(B. longum)[ASV73和ASV144]和假双歧杆菌(B. pseudocatenulatum)[ASV111和ASV153])、粪杆菌属(Faecalibacterium)和普拉梭菌(F.prausnitzii sp.)(ASV6、ASV10、ASV72和ASV95)、人型罗斯氏菌(Roseburia hominis)(ASV49和ASV188)和毛螺菌(Lachnospira sp.)(ASV23、ASV30和ASV37)均在恢复饮食期间得到富集(FDR调整后p < 0.05)。潜在促炎细菌类群的平均相对丰度降低,例如沃氏嗜胆菌(Bilophila wadsworthia)(ASV21)、腐败阿里斯提普斯菌(Alistipes putredinis)(ASV127和ASV171)、扭转地中海菌(Mediterraneibacter torques)(Ruminococcus torques的同义词;ASV167)和粪便副拟杆菌(Parabacteroides merdae)(ASV9、ASV130和ASV183)(图3B)。这些显著的改变得到了SGBs分析的进一步证明(表S5)。但恢饮食对于肠道菌群的影响及其效应大小仍然高度个性化,尽管这种影响在总体微生物类群层面上具有统计学意义,效应量也通常较大。例如,与基线相比,饮食引起消化道中双歧杆菌相对丰度变化从12%上升至760%不等,在七名受试者中,该属的相对丰度超过15%。此外,尽管在恢复饮食期间另枝菌属(Alistipes)和拟杆菌属(Bacteroides)总体上显著减少,但在约30%的受试者中,这些属的细菌数量有所增加(图S4)。恢复饮食干预下减少的许多肠道菌群分类单元被认为是BloSSUM分类单元(在城市化/现代化社会中被筛选或选择),而通常与非工业化人群相关的肠道菌群分类单元并没有因恢复饮食的干预而丰富,例如Segatella copri(数据未显示)。
恢复饮食改变了63种微生物途径(12种增加,51种减少)、95种ECs(36种增加,59种减少)和11种CAZymes(6种增加,5种减少)的平均相对丰度(FDR调整后p < 0.05,线性混合模型)(表S5)。恢复饮食增加了CAZymes的总丰度(p = 0.029,线性混合模型;图3D),尤其是针对植物碳水化合物利用的CAZymes(p = 0.021;图3E)。针对植物与动物碳水化合物的CAZyme比率从常规饮食中的1.50 ± 0.28增加到恢复饮食中的1.64 ± 0.30(p = 0.08;图3F),而粘蛋白与植物碳水化合物的比率从0.53±0.09降低到0.49±0.06(p=0.10;图3G)。属于GH43的四个CAZyme亚家族(GH43_22、GH43_26、GH43_27和GH43_29)在分解植物纤维(如半纤维素)中发挥作用,恢复饮食丰富了这些酶亚家族(FDR调整p < 0.05;图3H)。下降幅度最大的是CAZyme GH29家族(图3H),该家族中含有参与粘液降解的α-岩藻糖苷酶(alpha-fucosidases)。
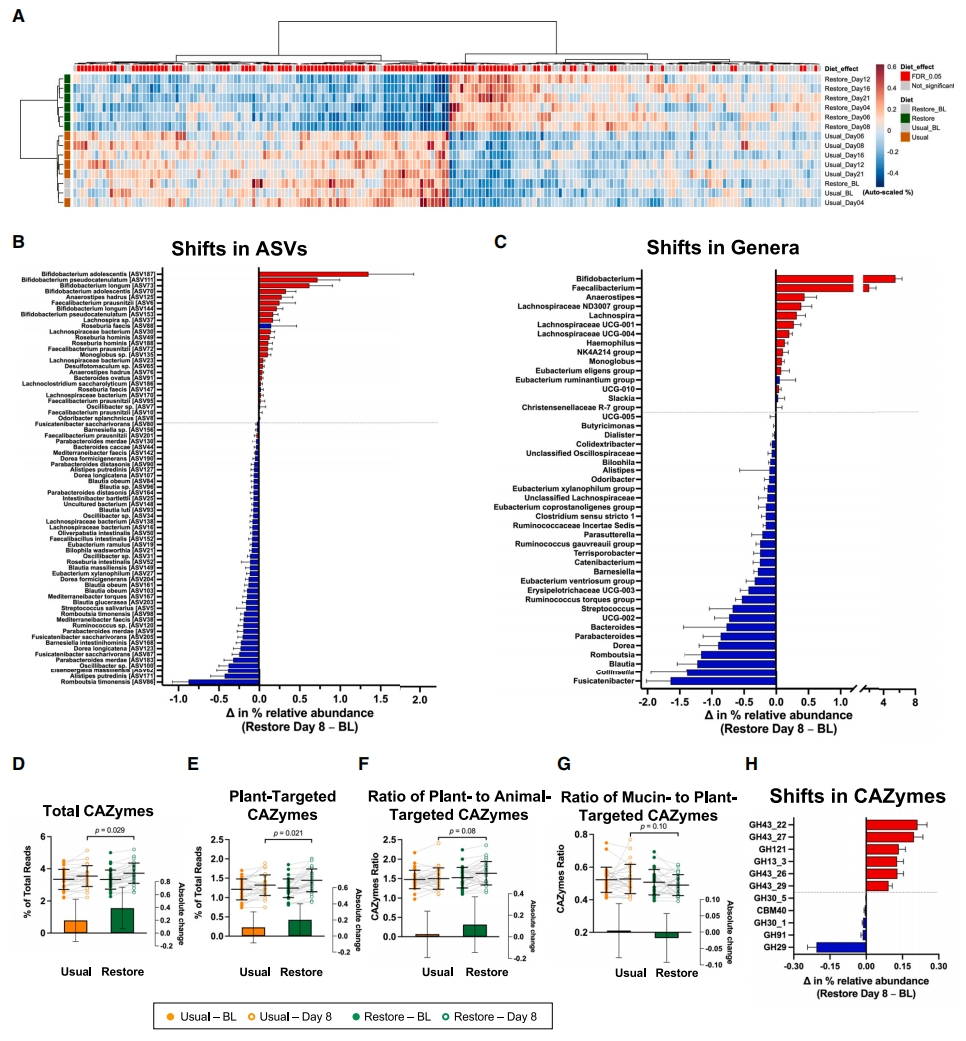
图3 | 恢复饮食改变的肠道微生物类群和CAZymes
(A–G)(A)热图显示每个采样日ASVs的平均相对丰度(基于使用欧几里得距离和Ward.D聚类的自动缩放相对丰度)。恢复饮食显著改变的ASVs(FDR调整p < 0.05;线性混合模型)以红色标识,而未受显著影响的ASVs以灰色标识。瀑布图显示显著改变的(B)ASVs和(C)属的相对丰度变化(线性混合模型;FDR调整p < 0.05)。根据系数着色的条形图-红色表示正,蓝色表示负。在每个饮食期间,CAZymes与不同碳水化合物来源的利用相对应的变化:(D)总CAZymes、(E)植物碳水化合物,以及(F)植物与动物碳水化合物和(G)粘蛋白与植物碳水化合物之间的比率(线性混合模型,FDR调整p < 0.05)。条形图(插图)表示每个饮食期间相对于基线值的绝对变化。
(H)瀑布图显示显著改变的CAZymes的相对丰度变化。数据以平均值 ± SD表示,符号代表单个样本。另见图S4和表S5。ASVs,扩增子测序变体;BL,每个饮食期的基线;CAZymes,碳水化合物活性酶。
饮食引起肠道微生物组扰动后的时间动态
我们利用交叉研究设计,并使用来自16S rRNA基因扩增子测序的数据集,分析摄入恢复饮食后一段时间内受试者的粪便样本(n = 16)中的微生物变化情况,以首先评估微生物群特征在何种程度上以及在哪个时间窗口期内恢复到基线水平。分析表明,alpha多样性指数(图4A和4B)、个体内beta多样性差异(图4C)和个体间beta多样性差异(图4D)在第一次洗脱期结束时或在正常饮食期开始后不久恢复到基线值。此外,受恢复饮食影响改变最显著的ASVs相对丰度也在第一次洗脱期后恢复到基线水平(图S5)。对于SGBs和WMS功能特征(微生物途径、CAZymes和ECs),也观察到了类似的恢复到基线值的情况(图S6A-6F)。总体而言,研究结果表明,恢复饮食对微生物组生态的影响是短暂且可逆的,并且微生物组对饮食变化表现一定程度的抗干扰性。
恢复饮食效果的生态驱动因素及其对微生物群落特征的影响
为了深入了解肠道微生物组变化的生态驱动因素,我们将多元线性回归(MLR)模型应用于第8天的微生物在属层面上的变化(16S rRNA基因扩增子测序),并在分析中涵盖了其他可能导致微生物组改变的因素——饮食、粪便pH值、粪便稠度和排便频率。该分析表明,大多数属不仅直接受到恢复饮食的影响(图S7A),而且拟杆菌属(Bacteroides)和副杆菌属(Parabacteroides)等属的减少与粪便pH值的变化有关(图S7B),这可能是由于它们对酸性pH值的敏感性。粪便稠度和排便频率仅影响少数属(图S7B),这与它们对整体微生物组成的影响有限的发现(图2K)相一致。
为了评估恢复饮食对微生物群落互联性的影响,我们使用线性模型比较了同一个体前后两个饮食期间纵向16S rRNA基因扩增测序中细菌在科水平上的相互作用网络,其中从时间点t到t+1的相对丰度变化由时间点t的丰度预测。该分析表明,在恢复饮食期间,正向和潜在的共生相互作用数量增多,负向相互作用保持不变(图4E和S7C)。毛螺菌科(Lachnospiraceae)、克里斯滕森菌科(Christensenellaceae)和瘤胃球菌科(Ruminococcaceae)等微生物科对其他几种分类群有显著的积极影响,因此被认为是恢复饮食期间与肠道微生物群调控相关的关键类群。在常规饮食中,毛螺菌科、瘤胃球菌科与其他分类群的相互作用大大减少,表明它们失去了在肠道菌群互作中的关键地位,这可能是因为在常规饮食中它们不需要发挥降解纤维素的能力。由于恢复饮食期间的正向相互作用数量较多,我们推测该微生物群会更加稳定。事实上,我们发现恢复饮食期间连续时间点之间的Bray-Curtis距离比正常饮食期间更小(p < 0.001,线性混合模型;图4F)。
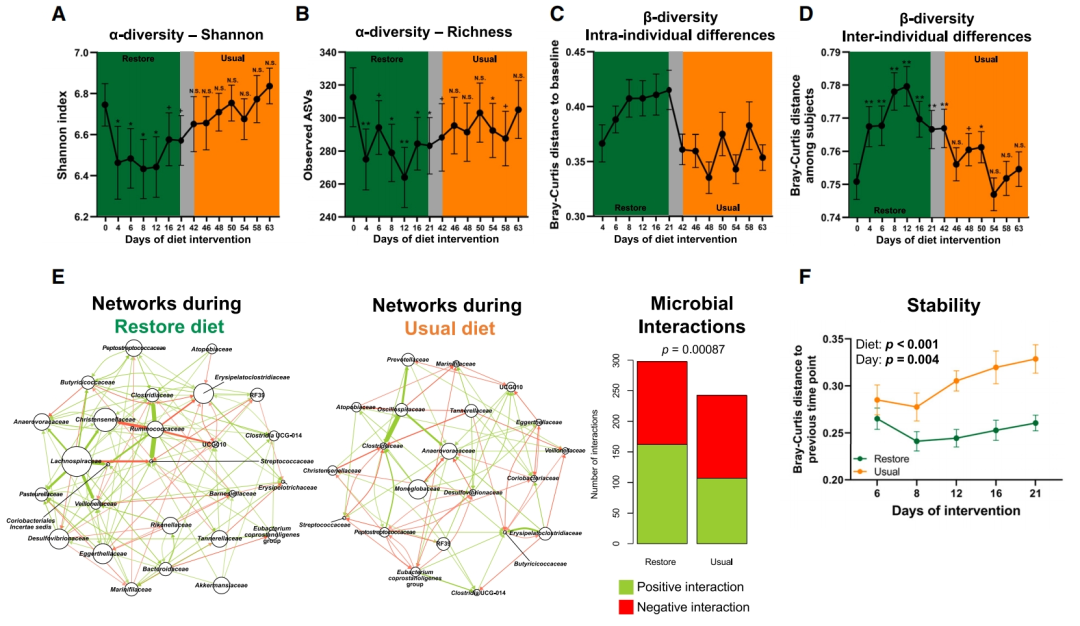
图4 | 饮食干预期间肠道微生物群的恢复力和稳定性以及这些生态反应的主要驱动因素
(A–D)食用恢复饮食后微生物群的时间响应和恢复力,表示为(A)Shannon指数(即alpha多样性)、(B)观察到的特征(即基于ASVs的丰富度)、(C)两个饮食期间每个受试者的微生物群与其基线相比的Bray-Curtis距离(即beta多样性,个体内距离)和(D)受试者微生物群之间的Bray-Curtis距离(即beta多样性,个体间距离)(配对Wilcoxon符号秩检验,p < 0.05)。数据来自16名随机首先食用恢复饮食的受试者。
(E)每个饮食期间细菌科的相互作用网络(线性模型)。节点大小表示节点分类单元影响的分类单元数量,箭头大小表示关联的估计值或强度(即分类单元的丰度与其他分类单元丰度变化的关联程度)。仅显示具有>2个相互作用且估计值为0.2的分类单元(即节点分类单元增加一个单位与其他分类单元丰度变化0.2个单位相关)。条形图表示微生物之间的正(绿色)和负(红色)相互作用的数量。
(F)肠道微生物群的时间变化,以两个饮食期间的两个连续时间点之间的Bray-Curtis距离表示(线性混合模型)。Bray-Curtis距离位于x轴上标记的“干预日”与前一个采样时间点之间,例如,“第6天”表示每个饮食期第4天和第6天之间的距离。数据以平均值 ± SEM表示。+p ≤ 0.1,*p ≤ 0.05,**p ≤ 0.001。另请参阅图S5–S7。ASVs,扩增子序列变体。
恢复饮食对微生物发酵和血浆代谢组的影响
恢复饮食使粪便pH值从6.82 ± 0.43降低至6.58 ± 0.44(p = 0.003;配对Wilcoxon符号秩检验;图5A),增加了总短链脂肪酸(SCFAs)(p = 0.028;图5B)和乙酸盐浓度(p = 0.023;图5C),降低了总支链脂肪酸(BCFAs)浓度(p = 0.045;图5D)和BCFAs与SCFAs的比率(p = 0.007;图5E)。总体而言,这些结果表明恢复饮食以减少蛋白水解代谢(BCFAs)为代价,增加了糖酵解发酵(SCFAs)(表S6)。
我们采用非靶向代谢组学分析血浆代谢物,并检测到528种高可信度代谢物。偏最小二乘判别分析(PLS-DA)表明,无论是恢复期(p = 0.72,图S8A)还是正常饮食期(p = 0.62,图S8B),三个接种组(PB-W1、DSM 20016T和安慰剂)的代谢物变化(给药后4天[第8天]-基线)没有显著差异。这些结果表明,单剂量罗伊氏乳杆菌不会影响血浆代谢组学特征。然而,当比较摄入恢复饮食(饮食干预结束[第21天]-基线)与常规饮食前后的各种代谢物时,PLS-DA揭示了由恢复饮食引起的整体代谢的的显著差异(p = 0.004;图5F)。
恢复饮食显著改变了31种代谢物的子集(FDR调整p < 0.10,配对t检验;图5G),其中22种增加(例如吲哚-3-丙酸、生物素、哌可酸)、9种减少(例如脱氧胆酸、8-羟基鸟嘌呤、熊去氧胆酸)。超过90%的代谢物是由微生物组产生或修饰的,或者由微生物组和宿主共同产生。相关性分析揭示了代谢物与肠道微生物属(图S8C)和ASVs(图S8D)之间的关系(FDR调整p < 0.10,Spearman秩相关性),这些关系因恢复饮食而显著改变,这表明肠道微生物组的变化导致了血浆代谢物的变化。例如,我们发现长双歧杆菌和吲哚-3-丙酸(图S8D)之间存在正相关性,而吲哚-3-丙酸是一种可由双歧杆菌产生的吲哚-3-乳酸合成的代谢物。
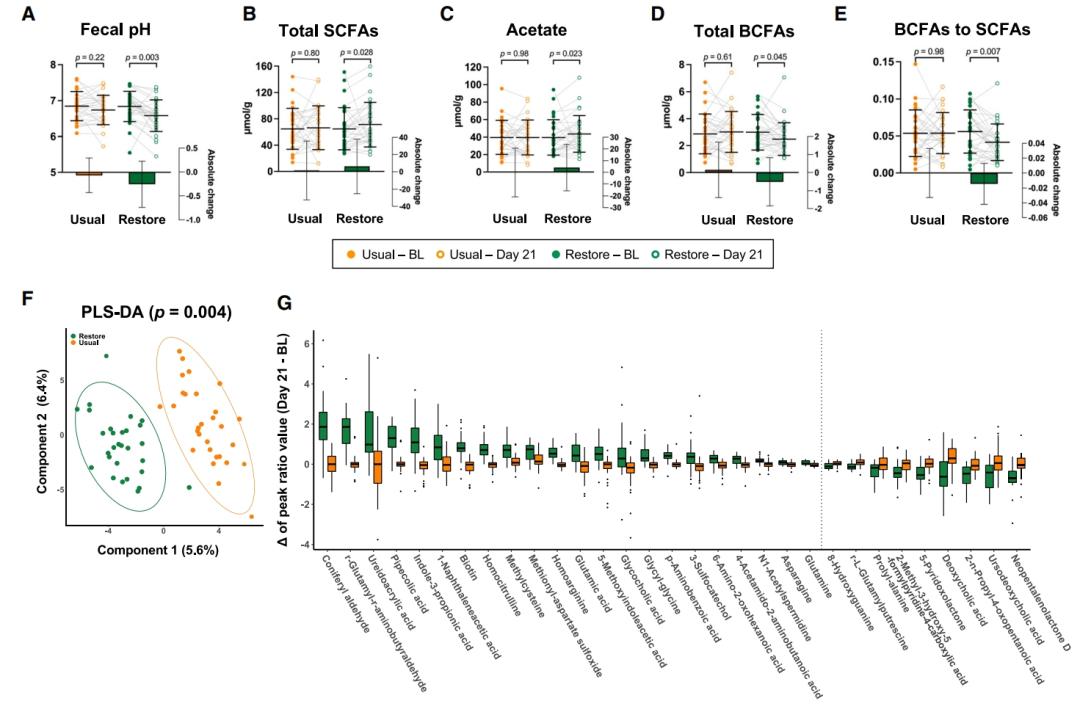
图5 | 恢复饮食对微生物发酵和血浆代谢组的影响
(A–E)每次饮食期间粪便(A)pH、(B)总SCFAs、(C)醋酸盐、(D)总BCFAs和(E)BCFAs与SCFAs比率的变化(配对Wilcoxon符号秩检验,p < 0.05)。数据以平均值 ± SD表示,符号代表单个样本。条形图(插图)表示每次饮食期间相对于基线值的绝对变化。
(F)从基线到每次饮食结束的血浆代谢组学谱变化(PLS-DA;p值来自1,000次置换验证)。
(G)从基线到每次饮食结束代谢物的变化(配对t检验,FDR调整p < 0.1)。左侧和右侧面板分别显示在恢复饮食期间增加和减少的代谢物。数据以箱线图表示,点代表异常值。另见图S7和S8以及表S6。
BCFAs,支链脂肪酸;BL,各饮食期的基线;PLS-DA,偏最小二乘判别分析;SCFAs,短链脂肪酸。
恢复饮食可显著改善慢性病风险指标
鉴于工业化和西方化饮食与非传染性疾病(NCDs)风险之间的联系,我们关注了微生物组恢复策略对NCDs风险标志物的影响。虽然罗伊氏乳杆菌不影响这些标志物(FDR调整后p > 0.05,线性混合模型;表S7),但恢复饮食具有明显的影响。风险标志物的主成分分析(PCA)排序显示每个饮食期的两基线之间没有差异(p = 0.96,PERMANOVA;图6A);然而,根据每个饮食期内标志物与基线水平相比的百分比变化,可以看出明显的聚类(p = 0.001;图6B),这表明恢复饮食引起了整体生理的显著变化。
尽管按照计算出的能量需求进食,受试者的体重还是出现了轻微但具有统计学差异的下降(从基线到恢复饮食第21天的百分比变化=1.4 ± 1.9%,平均值 ± SD;FDR调整后p = 0.003,线性混合模型;图6C)和BMI(1.4 ± 1.9%,p = 0.002;图6D)。这可能是由于富含纤维的植物性食物中大量营养素被三维细胞壁结构包裹,导致其生物可及性降低。
恢复饮食降低了空腹血浆总胆固醇(14.1 ± 11.2%,FDR调整后p < 0.001)、低密度脂蛋白(LDL)胆固醇(16.8 ± 15.8%,p = 0.003)、非高密度脂蛋白(非HDL)胆固醇(15.2 ± 14.5%,p = 0.002)、空腹血糖(6.3 ± 11.1%,p < 0.001)、C反应蛋白(CRP)(14.0 ± 58.3%,p = 0.014)和粪便钙卫蛋白(21.0 ± 88.3%,p = 0.011)(图6E;表S7)。胰岛素敏感性(定量胰岛素敏感性检查指数[QUICKI])增加(2.4 ± 6.4%,p = 0.036),胰岛素抵抗(胰岛素抵抗稳态模型评估[HOMA-IR])改善(5.8 ± 40.5%,p = 0.053),这主要是由于空腹血糖降低所致,因为对空腹胰岛素水平没有影响(p = 0.32)(表S7)。HDL胆固醇降低(11.3 ± 11.2%,p = 0.001),这在其他富含植物的饮食中也观察到了,而甘油三酯不受影响(p = 0.59)(表S7)。
当控制体重变化(数据未显示)时,恢复饮食的所有临床效果仍然具有统计学意义,但CRP除外(尽管p值保持在0.058的低位)。此外,当把饮食干预的顺序作为额外的协变量时(即:先恢复饮食再常规饮食 / 先常规饮食后恢复饮食),恢复饮食的效果并没有降低,这表明在两种饮食干预之间的3周洗脱期足以让受试者恢复到基线的代谢状态。考虑到恢复饮食中纤维摄入量的增加及其对发酵和SCFAs的影响,我们还关注了与肠道屏障功能相关的标志物是否受到影响。血浆脂多糖结合蛋白(8.1 ± 13.8%,p = 0.13)和粪便连蛋白(14.9 ± 27.6%,p = 0.052)均降低,但变化显著程度未达到统计学意义(表S7)。
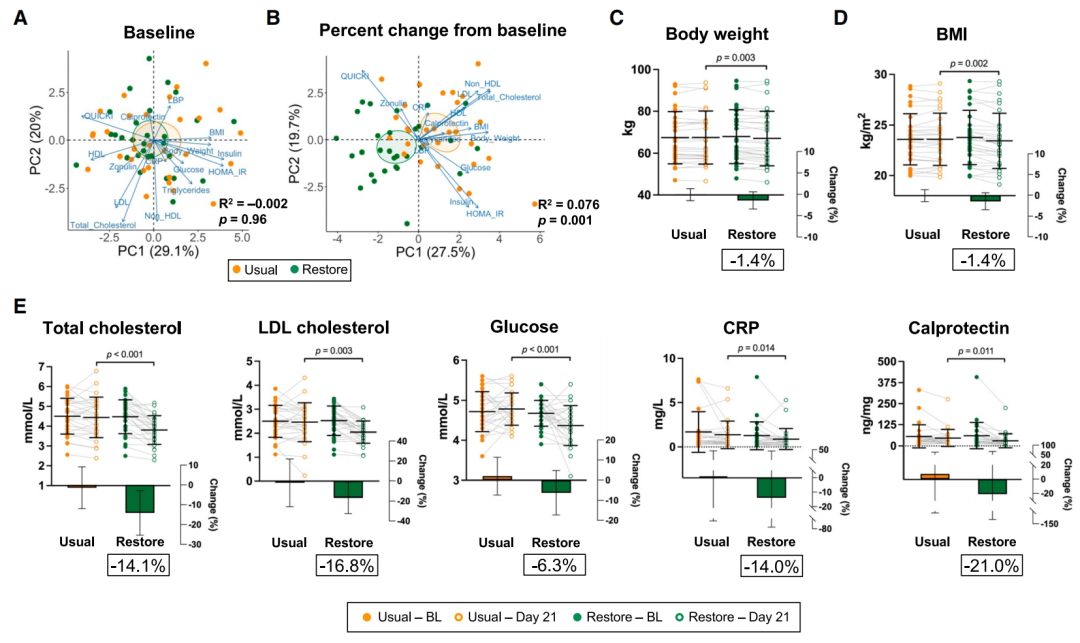
图6 | 恢复饮食对慢性病风险指标的影响
(A和B)PCA图和PERMANOVA结果显示(A)基线时的宿主参数(B)在每个饮食期间相对于基线的百分比变化。
(C–E)每个饮食期间风险标记的差异(线性混合模型,FDR调整p < 0.05)。数据以平均值 ± SD表示,符号代表单个样本。条形图(插图)表示每个饮食期间相对于基线值的百分比变化。恢复饮食中的平均百分比变化在每个图下方的文本框中。BL,每个饮食期的基线;BMI,身体质量指数;HDL,高密度脂蛋白;HOMA-IR,胰岛素抵抗的稳态模型评估;CRP,C反应蛋白;LBP,脂多糖结合蛋白;LDL,低密度脂蛋白;QUICKI,定量胰岛素敏感性检查指数。另见表S7。
鉴定与临床结果相关的微生物组和代谢组特征
为了获得对恢复饮食生理效应机制的解释,我们采用了一组互补方法,使用机器学习(随机森林[RF])和MLR模型将临床发现与肠道微生物组和血浆代谢组的数据相结合。我们将这些分析应用于单个组学数据集(即血浆代谢物、多个分类学水平的微生物组组成概况、推断的微生物组功能、SCFAs和肠道微生物组的多样性指数),目的是确定饮食对生理效应作用的最佳预测因子,同时使用基线特征和饮食引起的绝对变化。基线饮食摄入量和代谢状态(即基线风险标记,如空腹血糖、胰岛素、脂质组等)也被考虑在内(参见STAR方法)。
作为概念验证,我们首先判定饮食引起的微生物组、代谢组和代谢状态的变化是否可以预测受试者是在摄入常规饮食还是恢复饮食。利用在每个饮食期结束时的血浆代谢物(受试者工作特征曲线下平均面积[AUROC] = 0.99)和每个饮食期第8天的基于WMS的微生物群组成概况(AUROC = 0.96),以及饮食引起的血浆代谢物变化(AUROC = 0.99)和基于WMS的微生物群组成概况(AUROC = 1.00)(图S9B;表S8),RF分类模型在判定的准确度上几乎完美(图S9A;表S8)。总体而言,WMS衍生的功能特征(即CAZymes、通路和ECs)也是很好的预测因子(AUROC0.75–0.91),但SCFAs并不优于随机概率(AUROC 0.48和0.62)(图S9A和S9B;表S8)。这些结果表明,恢复饮食对微生物组和代谢组的影响足够强大和具体,可以几乎完美地预测受试者的饮食状况。
唯一对恢复饮食显示出个体化反应的风险标记是血浆葡萄糖(图6E),有明确的反应者和无反应者。使用RF分类模型预测“反应者”(葡萄糖降低≥ 0.3mmol/L)和“无反应者”(降低< 0.3mmol/L),基于基线代谢状态的模型准确预测了血糖反应(AUROC = 0.82;图7A;表S9)。首要的预测特征是基线CRP、葡萄糖和HOMA-IR水平,这些水平在“反应者”中升高,这表明受试者的基线免疫代谢状态(例如炎症和血糖升高)决定了对恢复饮食的反应。尽管该模型的准确度很高且预测因子具有生物学意义,但使用基于基线微生物组组成(ASVs水平,AUROC = 0.92;图7A;表S9)、微生物途径变化(AUROC = 0.94)和血浆代谢物(AUROC = 0.84)搭建的RF模型甚至更为准确(图7B;表S10)。值得一提的是,最主要的预测途径——四吡咯生物合成I(来自谷氨酸)和代谢物1-吡咯啉-2-羧酸——都与吡咯代谢有关。这些预测因子的变化彼此呈负相关(rs = 0.52,FDR调整后的p = 0.06)(图7C),并且与葡萄糖的百分比变化(四吡咯生物合成I[来自谷氨酸]rs = 0.45,图7D)呈现相反的关联性。1-吡咯啉-2-羧酸可以从脯氨酸合成,脯氨酸是一种可以通过糖异生转化为葡萄糖的氨基酸。因此,在响应者中,通过四吡咯生物合成I途径由微生物组驱动的1-吡咯啉-2-羧酸减少可能通过减少脯氨酸来降低葡萄糖浓度。这些发现表明,微生物组-宿主代谢串扰在恢复饮食对血浆葡萄糖浓度的影响中起着作用。MLR证实血浆代谢物是葡萄糖变化的预测因子(R2 = 0.23,p = 0.021)(表S11)。
与空腹血糖相比,恢复饮食对其他风险标志物的影响在不同受试者间差异非常小(图6E)。因此,我们应用RF回归模型和MLR来评估相关临床反应在多大程度上可以通过微生物组、代谢组或其他宿主特征来解释,并确定了最佳预测因子。基线微生物组组成(科水平)解释了不同受试个体间LDL变化的大幅差异(RF平均R2 =0.75;MLR R2 = 0.29,p = 0.010)(图7E;表S9和S11)。基线粪便SCFA水平是CRP水平变化的良好预测指标(RF平均值R2 = 0.47;MLR R2 = 0.35,p = 0.012)(图7E;表S9和S11),BCFA与SCFA的比率以及丙酸和总SCFA的浓度可作为模型的主要的预测特征。有趣的是,高比率的BCFA、SCFA(rs = 0.62,p = 0.001)和低水平的丙酸、总SCFA(分别对应:rs = 0.60,p = 0.002和rs = 0.54,p = 0.005)与CRP的大幅下降相关(图7E)。这表明,如果饮食调控前宿主肠道微生物群的发酵情况不佳(即蛋白质较多而纤维发酵较少),恢复饮食的抗炎效果会更好,但因果关系尚无法阐明(例如,基线微生物组代谢可能会影响临床反应,但也可能受到独立因素的影响)。与葡萄糖反应不同,基线宿主代谢状态无法解释其他风险标志物的大量变化(表S9)。
MLR分析显示,总胆固醇(R2 = 0.31,p = 0.007)、LDL(R2 = 0.28,p =0.011)和非HDL(R2 = 0.24,p = 0.021)的变化可以通过基线微生物组多样性指标(表S11)来解释,第二主成分(PC2;占总变异的27.0%)与这些风险标志物呈负相关(数据未显示)。主要变量(解释该PC内>10%的变异)是观察到的CAZymes特征(12.71%)、SGBs的Shannon指数(12.05%)和SGBs的Pielou均匀度(10.80%)。总而言之,该分析表明,基线状态下肠道微生物群更高的多样性指数与胆固醇标志物的更明显的降低相对应。虽然我们仅使用MLR检测到基线微生物组多样性指标与CRP之间的趋势(p = 0.086;表S11),但我们的研究结果在概念上与先前的研究结果一致,该研究表明,肠道微生物组多样性较高的受试者对高纤维饮食的免疫反应更好。
当使用饮食引起的变化构建模型时,属级的微生物组成产生了预测LDL变化的最佳模型(RF平均值R2 = 0.66),其中最佳预测因子是与LDL变化呈正相关的嗜胆菌属(Bilophila)变化(意味着饮食引起的嗜胆菌属减少与LDL减少有关)(rs = 0.54,p = 0.027)(图7F;表S10)。CAZyme比率的变化产生了CRP的最佳预测模型(RF平均值R2 = 0.44;MLR R2 = 0.34,p = 0.010),其中针对粘蛋白聚糖和动物碳水化合物的CAZymes丰度与总读数之比是最重要的预测因子(图7F;表S10和S11)。这两个特征与CRP的变化呈正相关(rs = 0.51和0.43,p = 0.012和0.028),表明与粘蛋白聚糖和动物碳水化合物代谢相关的CAZymes丰度的大幅下降与CRP的大幅下降相关。
为了使用独立的方法佐证微生物组特征与宿主临床反应之间的联系,我们利用逐步简化上述模型的方法建立了线性回归模型。该分析表明,可通过微生物组组成和功能变化(变化最显著的因素)来高精度地预测恢复饮食的临床反应(图S10A)。某些变量与RF分析中确定的最佳预测因子一致(图7A和7F),例如嗜胆菌属的减少和更高的基线葡萄糖水平,分别对应LDL和葡萄糖更大程度的降低(图S10B)。该分析确定了独立于微生物组的恢复饮食对LDL、钙卫蛋白和葡萄糖的可测量的影响。相比之下,恢复饮食对总胆固醇、CRP和BMI仅具有独立于微生物组的,轻微的直接影响,说明这些参数的降低主要是受微生物组的反应驱动的。
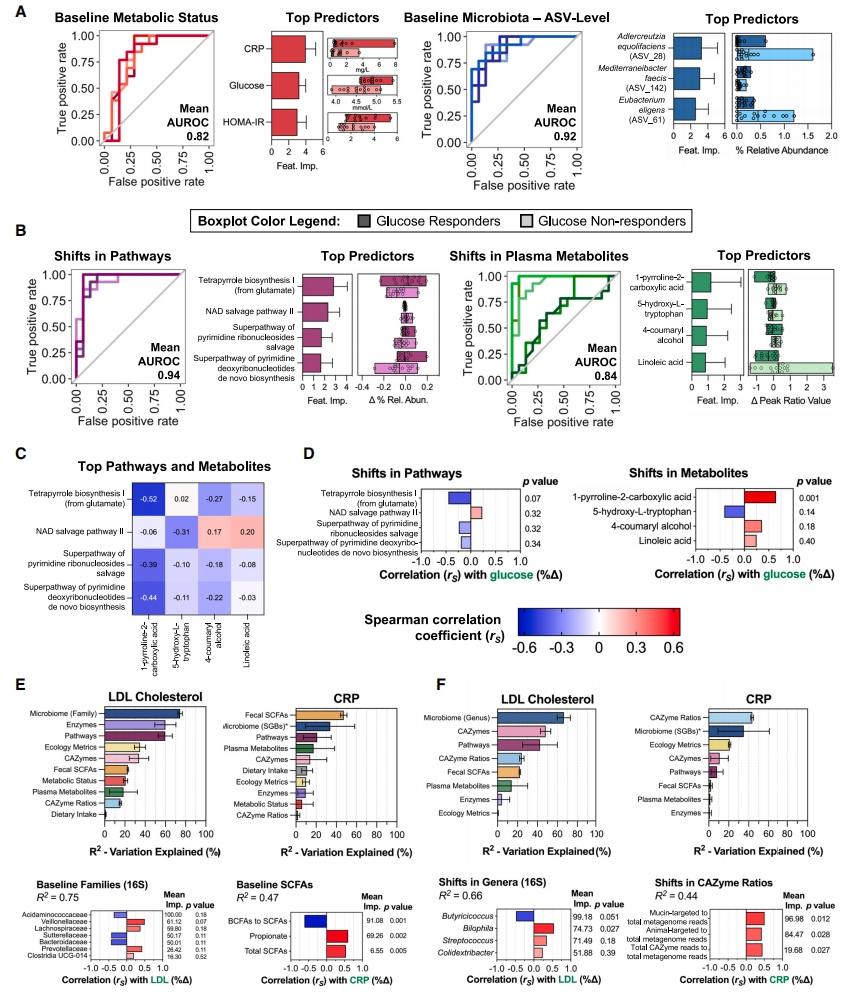
图7 | 数据整合以确定预测慢性疾病风险标志物对恢复饮食反应的变量
(A和B)使用RF分类模型预测有反应者(葡萄糖R降低0.3mmol/L)和无反应者(降低< 0.3mmol/L),使用(A)基线值(最佳模型:代谢状态和微生物组成、ASV水平)和(B)特征的绝对变化(最佳模型:通路和血浆代谢物)。模型性能以ROC曲线表示-图包含五条单独的曲线,每条曲线代表5倍外部交叉验证之一的ROC。使用不同深浅的主色,使重叠线清晰可见,灰线表示AUROC为0.5(即不比随机猜测好)。最佳模型的顶级预测特征可视化为具有平均值± SD特征重要性的条形图,箱线图显示有反应者和无反应者之间这些特征的差异。
(C)热图显示顶级预测途径和代谢物之间的Spearman相关系数(rs)。
(D–F)(D)条形图显示葡萄糖百分比变化与顶级预测途径和代谢物之间的相关性(rs)。使用(E)基线值和(F)特征的绝对变化对LDL胆固醇和CRP应用RF回归模型。模型性能表示为R2(解释的变化比例)。水平条显示平均R2,误差条为95%置信区间。*表示通过WMS评估的微生物组组成。没有*表示通过16SrRNA基因扩增子测序评估的微生物组组成。仅显示表现最佳的微生物组组成模型以避免重复。预测LDL胆固醇和CRP百分比变化的最佳模型的最佳预测因子与(D)中的类似。FDR调整后的p < 0.05。另请参见图S9–S11和表S8、S9、S10和S11。ASVs,扩增子序列变体;AUROC,受试者工作特征曲线下面积;BCFAs,支链脂肪酸;CAZymes,碳水化合物活性酶;CRP,C反应蛋白;ROC,受试者工作特征;SCFAs,短链脂肪酸。
作者简介
浙江大学动物科学学院李福勇研究员和阿尔伯塔大学医学院儿科系Anissa Armet博士是本文的第一作者,爱尔兰APC微生物组研究所和科克大学消化道微生物进化生态学教授Jens Walter为本文的通讯作者。
李福勇(第一作者)

浙江大学第一类“百人计划”研究员,博士生导师,国家高层次青年引进人才,主持国家自然科学基金海外高层次引进人才青年项目和面上项目。长期致力于消化道功能微生物组这一国际前沿领域的研究,结合组学技术、分子微生物学技术及经典微生物学技术,阐释了消化道微生物与宿主在营养和遗传层面的交互作用,揭示了消化道微生物对于宿主的适应性进化模式,取得了一系列创新性学术成果。共发表SCI论文36篇;其中以第一或通讯作者在Cell、Microbiome(3篇)、iMeta等杂志发表论文12篇,包括高被引论文1篇、杂志亮点文章1篇。现担任Microbiome杂志副主编。
Anissa Armet(第一作者)

注册营养师,于阿尔伯塔大学农业、食品与营养科学系取得博士学位,现任职于阿尔伯塔大学医学院儿科系从事博士后研究。Anissa Armet博士的研究聚焦于通过营养策略调控肠道微生物群,从而促进人类健康。具体而言,其研究兴趣涵盖个性化与精准营养领域,以及基于肠道微生物组预测营养干预措施对人类健康的影响。目前已在Cell、Cell Host & Microbe、Microbiome等期刊发表论文多篇。
Jens Walter(通讯作者)

现任爱尔兰APC微生物组研究所和科克大学消化道微生物进化生态学教授,曾先后任职于美国University of Nebraska–Lincoln、加拿大University of Alberta等国际知名高校。他聚焦于探究塑造宿主-微生物共生关系的进化和生态机制,旨在将基础微生物组学研究转化为营养策略制定。Jens Walter教授曾先后主持多项声誉卓越的基金项目,包括加拿大卫生研究院项目、加拿大自然科学与工程研究理事会项目、NIH项目以及多项国际合作项目。至今共发表SCI期刊论文200余篇(H-index 86),在Cell、Nature Reviews Microbiology、Nature Microbiology、Cell Host & Microbe等顶级期刊发表多篇重要著作。自2019年起,连续多年入选Web of Science高被引研究者。
翻译:马闯,安徽农业大学,基因组所客座硕士在读
审核:李福勇,浙江大学,研究员/博导
排版:杨海飞,青岛农业大学,基因组所联培硕士在读
宏基因组推荐
本公众号现全面开放投稿,希望文章作者讲出自己的科研故事,分享论文的精华与亮点。投稿请联系小编(微信号:yongxinliu 或 meta-genomics)
猜你喜欢
iMeta高引文章 fastp 复杂热图 ggtree 绘图imageGP 网络iNAP
iMeta网页工具 代谢组MetOrigin 美吉云乳酸化预测DeepKla
iMeta综述 肠菌菌群 植物菌群 口腔菌群 蛋白质结构预测
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature
一文读懂:宏基因组 寄生虫益处 进化树 必备技能:提问 搜索 Endnote
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流快速解决科研困难,我们建立了“宏基因组”讨论群,己有国内外6000+ 科研人员加入。请添加主编微信meta-genomics带你入群,务必备注“姓名-单位-研究方向-职称/年级”。高级职称请注明身份,另有海内外微生物PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。
点击阅读原文


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









