






点击蓝字 关注我们
棉花抗黄萎病的全景解析:从遗传基础到精准基因组选择

iMeta主页:http://www.imeta.science
研究论文
● 原文: iMeta (IF 23.8)
● 原文链接: https://onlinelibrary.wiley.com/doi/full/10.1002/imt2.70029
● DOI: https://doi.org/10.1002/imt2.70029
● 2025年4月11日,华中农业大学朱龙付、张献龙和金双侠等在iMeta在线发表了题为“A panoramic view of cotton resistance to Verticillium dahliae: from genetic architectures to precision genomic selection”的文章。
● 本研究系统解析了棉花抗黄萎病的遗传基础与调控网络,阐明了棉花抗黄萎病育种改良的遗传学机制。通过多环境鉴定的10个稳定QTL,构建了高效的棉花抗黄萎病基因组选择育种模型,并利用F2:3群体验证了该模型对棉花抗黄萎病表型的预测能力,为基于基因组选择的棉花抗病育种提供了新策略。整合基因组、群体转录组与功能基因组学分析,揭示了活性氧(ROS)在棉花抗黄萎病中的核心调控作用。研究成果为棉花抗病育种提供了宝贵的遗传资源,并为解析棉花及其他作物抗黄萎病的遗传基础和调控机制提供了重要理论依据。
● 第一作者:张晓君、柳仕明
● 通讯作者:朱龙付(lfzhu@mail.hzau.edu.cn)、张献龙(xlzhang@mail.hzau.edu.cn)、金双侠(jsx@mail.hzau.edu.cn)
● 合作作者:吴鹏、许琬莹、杨定宜、明雨晴、肖胜华、王为然、马军、聂新辉、高瞻、吕俊元、武斐、杨兆光、郑宝鑫、杜萍、王江媚、丁昊、孔杰、阿里甫·艾尔西、余渝、高巍、林忠旭、尤春源、Keith Lindsey、Nataša Štajner、王茂军、吴家和
● 主要单位:华中农业大学、新疆农科院、新疆农垦科学院、石河子大学等
亮 点
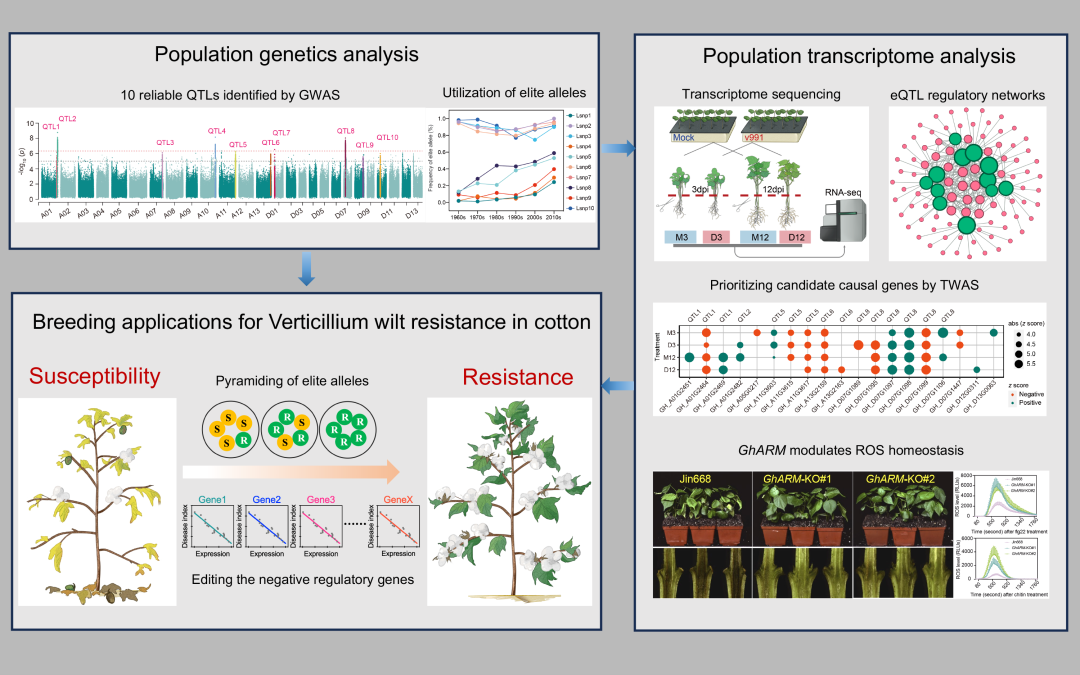
● 基于290份陆地棉种质的GWAS分析,鉴定出10个多环境稳定的棉花抗大丽轮枝菌QTL。10个优异等位基因的聚合使棉花黄萎病病指从70降至20;
● 优异等位基因的历史演化分析表明,中、低频率的Lsnp1R、Lsnp4R、Lsnp5R、Lsnp8R和Lsnp9R自20世纪90年代以来的利用频率演变与中国棉花育种进程吻合,为解析中国抗黄萎病育种进展提供新视角;
● 通过正常与胁迫条件下1152份转录组的分析,揭示棉花响应大丽轮枝菌的调控网络,突出ROS稳态与免疫应答的核心作用;
● GWAS与TWAS联合分析锁定15个候选因果基因,其中GhARM通过调节ROS稳态负调控棉花对黄萎病的抗性。
摘 要
解析复杂性状的遗传调控机制是作物改良的基础。由大丽轮枝菌(Verticillium dahliae)引起的黄萎病(Verticillium wilt, VW)是全球作物生产中最具破坏性的病害之一。然而,作物抗大丽轮枝菌的遗传基础仍不明确,阻碍了抗黄萎病基因组选择育种的进程。本研究通过整合全基因组关联分析(GWAS)和转录组关联分析(TWAS),利用290份陆地棉种质产生的1152份转录组数据,系统解析了棉花抗黄萎病的遗传基础与调控网络。我们在多环境中鉴定到10个与抗黄萎病相关的可靠数量性状位点(QTL)。这些QTL表现出显著的聚合效应,基于此开发了棉花对黄萎病的病指预测模型,并利用F2:3群体验证了其在棉花抗黄萎病基因组预测中的效果。此外,优异等位基因的历史演化分析表明,Lsnp1R、Lsnp4R、Lsnp5R、Lsnp8R和Lsnp9R的利用频率自20世纪90年代以来显著增加,可能推动了中国棉花抗黄萎病育种的改良。研究还发现了与活性氧(ROS)稳态调控及免疫应答相关的核心基因模块和表达QTL(eQTL)热点。通过TWAS进一步筛选出15个候选因果基因,沉默8个负调控基因显著增强了棉花对大丽轮枝菌的抗性。特别地,编码ARM重复蛋白的GhARM被证实通过调控ROS稳态介导棉花抗病性。本研究深化了对棉花抗黄萎病遗传基础与调控机制的理解,为基于基因组选择的抗病育种提供了新策略。
视频解读
Bilibili:https://www.bilibili.com/video/BV1sXdzYKEfF/
Youtube:https://youtu.be/4fqLG4lzNS4
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
棉花是全球重要的经济作物,为纺织工业提供35%的天然纤维。由大丽轮枝菌(Verticillium dahliae)引起的黄萎病(Verticillium wilt,VW)是一种毁灭性土传病害,可侵染棉花、番茄、马铃薯、橄榄和啤酒花等200余种植物,导致作物减产10%−60%,并造成严重经济损失。黄萎病每年导致全球棉花产量损失10%−35%,在中国,40%以上的棉田受其影响,年均经济损失达2.5−3.1亿美元。此外,大丽轮枝菌以微菌核形式在土壤中长期存活积累,秸秆还田和连作的耕作方式导致棉花黄萎病发病率逐年攀升。培育抗病品种是防控该病害最有效的策略。
遗传复杂性是数量性状改良的核心制约因素,尤其在植物抗病性中,单一抗病基因易被快速进化的病原菌所突破。遗传解析表明,番茄对1号生理小种(race1)的抗性由显性基因Ve1控制,而棉花、茄子和马铃薯的抗性则表现为典型数量性状遗传。通过全基因组关联分析(GWAS),研究者在A10和D11染色体上定位到了黄萎病抗性相关位点。病毒诱导基因沉默(VIGS)技术也鉴定了若干调控棉花抗性的基因,如编码NBS-LRR蛋白的CG02、编码Toll/白介素1受体(TIR)NLR(TNL)的GhRVD1以及编码(S)-去甲乌药碱合酶的GhNCS。然而,针对棉花抗大丽轮枝菌这一复杂数量性状,全基因组水平的抗病位点鉴定与解析对抗病育种至关重要。
人类基因型-组织表达(GTEx)计划系统解析了遗传变异对多组织基因表达的调控。研究者可通过转录组关联分析(TWAS)整合GTEx的表达数量性状位点(eQTL)与GWAS数据,解析复杂疾病的调控机制。近年来,eQTL与TWAS分析已拓展至作物研究:玉米中用于解析耐旱、耐热和温带适应等复杂农艺性状的遗传调控;甘蓝型油菜中聚焦种子含油量;水稻中用于探究穗型结构和耐盐性。在棉花中的研究主要集中于纤维品质,也有针对高温胁迫和棉籽产量的报道,但在棉花抗大丽轮枝菌的遗传调控机制中缺乏相关的应用。TWAS通过优先筛选GWAS区间内与目标性状关联的候选基因,为遗传调控网络解析提供新视角。
过去50年,全球围绕棉花抗大丽轮枝菌的遗传基础与育种开展了持续研究,但棉花品种的抗性提升效果不佳,选育高抗品种仍是重大挑战。为系统解析棉花抗黄萎病的遗传基础与调控机制,推动抗病基因组选择育种,本研究整合GWAS、TWAS与基因共表达网络分析,基于290份棉花种质的基因组变异图谱及正常/胁迫条件下的1152份转录组数据,鉴定抗性相关QTL、候选基因及调控网络。通过基因沉默和敲除实验验证多组学筛选的候选基因功能,揭示其抗病机制。该研究为棉花抗黄萎病遗传改良与复杂性状基因组选择提供了理论依据。
结 果
GWAS解析陆地棉抗大丽轮枝菌的复杂遗传基础
本研究利用已重测序的290份陆地棉自然群体(表S1),连续三年在新疆玛纳斯、库尔勒和库车的黄萎病病圃进行表型鉴定,获得5个独立环境(18-玛纳斯、18-库尔勒、18-库车、19-库车和20-库车)的9组原始病指(DI)(图1A和S1)。原始DI频数呈正态分布(图S2)。通过最佳线性无偏估计(BLUE)计算获得7组BLUE DI,与原始DI高度相关(图1A和S3)。基于双重复最大值合并DI数据集,获得4组合并DI,共生成20组DI数据(图1A和表S2)。同地点皮尔逊相关系数(r = 0.33−0.67)高于同年份(r = 0.13−0.63)(图1B),表明环境差异是抗性表型变异的主要因素。对于5个独立环境的DI数据,基于Kmeans方法将290份种质聚为5类(图1C和S4),其中78份(27.4%)在所有环境中均感病,14份(4.9%)在所有环境中抗病,67.7%的种质抗性不稳定(图1C),该结果表明环境对棉花黄萎病的发生具有显著影响,且我国陆地棉种质中缺乏稳定的高抗品种。
基于GEMMA混合线性模型,利用2,152,153个SNP对20组DI进行GWAS分析(图S5),共鉴定到22个显著关联位点,分布于15条染色体(At亚基因组13个,Dt亚基因组9个)(图1D和表S3)。其中10个位点在3个及以上的DI数据集中重复检测出(6个环境稳定位点,4个环境特异位点),将其定义为棉花抗黄萎病QTL(图1D、表1和S3)。每个QTL中p值最显著的SNP命名为Lead SNP(Lsnp),每个Lsnp的表型贡献率为7.21%−12.36%,与DI显著相关(图1E,F)。
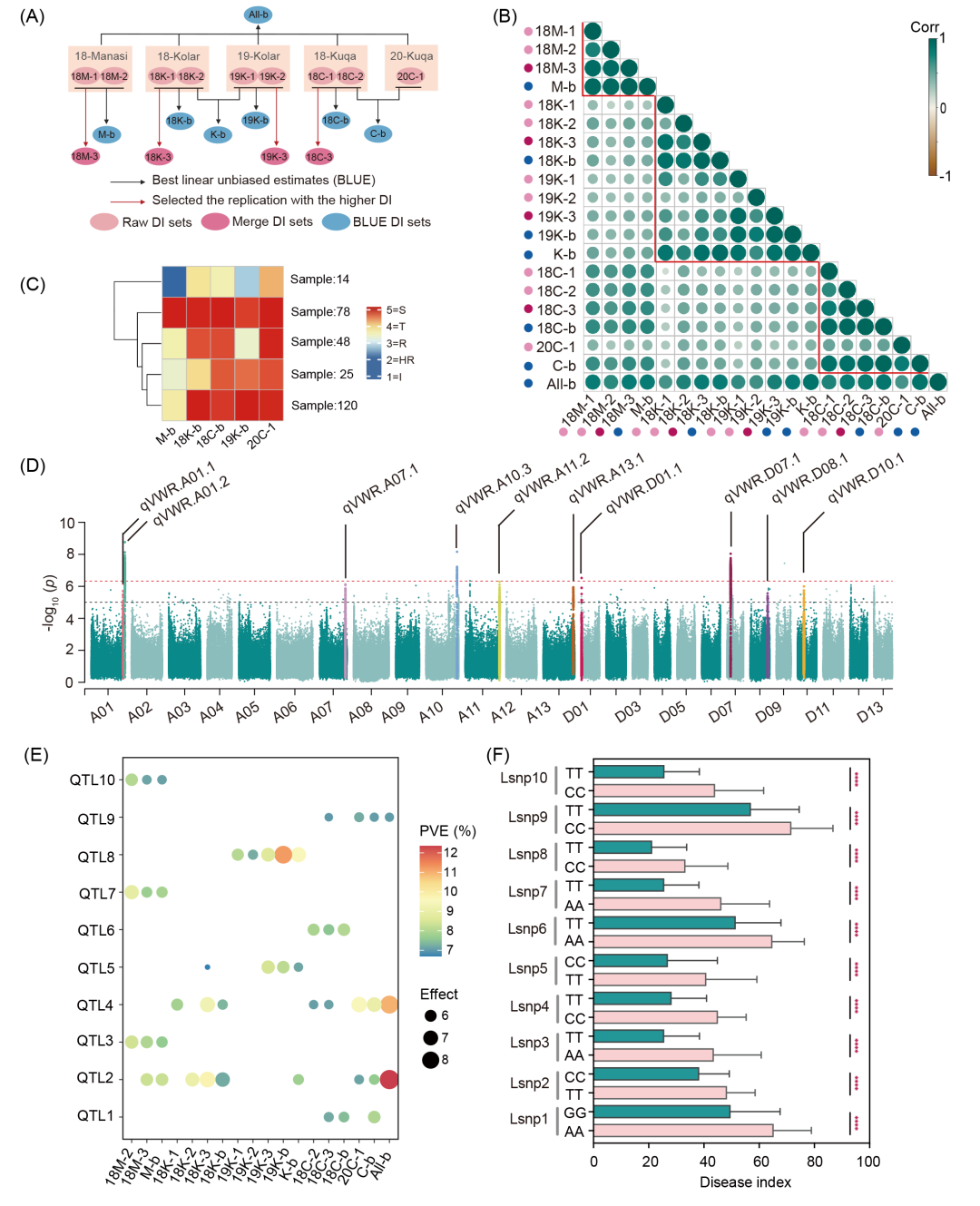
图1. 不同环境下棉花抗黄萎病表型分析与全基因组关联分析
(A) 病情指数(DI)数据处理流程示意图。粉色、品红色与蓝色圆圈分别代表原始DI、合并DI与BLUE DI数据集。命名规则为:年份-地点-数据类型(18/19/20对应2018/2019/2020年;M/K/C分别代表玛纳斯、库尔勒、库车;后缀1/2/3/b分别表示重复1、重复2、合并DI与BLUE DI);(B) 20组DI数据集相关性分析。圆圈大小表示皮尔逊相关系数;(C) 五组独立环境下DI聚类热图。色阶1-5分别对应免疫(I)、高抗(HR)、抗病(R)、耐病(T)与感病(S)。样本数标注于聚类分支;(D) 20组DI的GWAS曼哈顿图。标注为重复出现 ≥ 3次的QTL。横轴为陆地棉26条染色体(A01−A13,D01−D13),纵轴为−log10 (p)。显著性阈值:经验阈值p = 1E-05(虚线)、Bonferroni校正阈值p = 4.65E-05(实线);(E) 10个QTL的表型解释率(PVE)与效应值。圆圈大小表示效应值,颜色深浅表示PVE百分比;(F) 10个QTL 的Lsnp不同基因型对应的DI分布。双尾t检验,****p < 0.0001。柱高代表均值,误差线为±标准差。
中国栽培品种中10个LsnpRs的遗传结构解析
中国早期陆地棉种质资源主要来自于20世纪的国外引种材料,包括美国(如金字棉、德字棉531C、岱字棉15、斯字棉2B及脱字棉)、前苏联(如611B、KK1543、108夫及C1470)和乌干达(如乌干达3号及乌干达4号),这些品种构成了我国棉花育种的祖先种质。为解析我国抗病育种的历史与成效,本研究在11份祖先种质中分析了10个Lsnp的情况(图2A)。结果显示,10个LsnpRs(LsnpR代表Lsnp的抗性等位基因)可分为三类:美/苏/乌共有型(ASU)、苏/乌共有型(SU)及其他型(图2A)。其中Lsnp2R、Lsnp3R、Lsnp5R、Lsnp6R、Lsnp7R、Lsnp8R和Lsnp10R存在于ASU祖先种质,Lsnp9R仅存于SU种质(图2A),而Lsnp1R和Lsnp4R未在祖先种质中出现,推测来源于其他种质或育种过程(图2A)。为解析这10个LsnpR在我国棉花品种中的遗传特征,我们收集了2033份中国棉花栽培种(CDCs),其中1006份具有审定年份信息,1885份具有地理分布信息(图S6A,B和表S4)。结果表明,祖先种质中常见的Lsnp2R、Lsnp3R、Lsnp6R、Lsnp7R和Lsnp10R在CDCs中也保持高利用频率(HF)(图2A,B);祖先种质中常见的Lsnp8R与稀有的Lsnp5R在CDCs中的频率相当,为中等频率(MF)(图2A,B);祖先种质中缺乏的Lsnp1R、Lsnp4R和Lsnp9R在CDCs中呈现低频率(LF),分别为8.39%、8.33%和16.42%(图2A,B),该结果表明这些抗病等位基因尚未被充分利用,也暗示了我国棉花抗病育种中引入的抗源有限。
本研究进一步分析了10个LsnpR在CDCs中利用频率的历史演化规律。高频LsnpR(Lsnp2R、Lsnp3R、Lsnp6R、Lsnp7R和Lsnp10R)在历史审定品种中持续保持73.91%−100%的高利用频率(图2C);中、低频的Lsnp1R、Lsnp4R、Lsnp5R、Lsnp8R和Lsnp9R的利用频率自20世纪60年代至21世纪10年代增长超3倍,其中中频的Lsnp5R和Lsnp8R从17.76%(1960s)升至58.82%(2010s),而低频的Lsnp1R、Lsnp4R和Lsnp9R在2010年后的品种中仍低于40%(图2C)。值得注意的是,中、低频LsnpR的频率自20世纪90年代起显著提升(图2C,D)。地理分布分析显示,1885份CDCs可划分为四大棉区:辽河流域棉区(LRR,71份)、黄河流域棉区(YRR,946份)、长江流域棉区(YZR,527份)和西北内陆棉区(NIR,341份)(图S6B)。中频Lsnp5R和Lsnp8R在四大植棉区呈现最大变异度,其中长江流域频率最低(图2E)。总体而言,LsnpR的利用频率以辽河流域品种最高(59.7%),长江流域品种最低(52.21%)(图2E),表明辽河流域和黄河流域的品种对大丽轮枝菌具有更高的抗性。长江流域的水旱轮作模式限制了病原菌持续积累,而辽河流域和黄河流域的品种因病原菌选择压力更大,表现出更强的抗病性(图S7)。
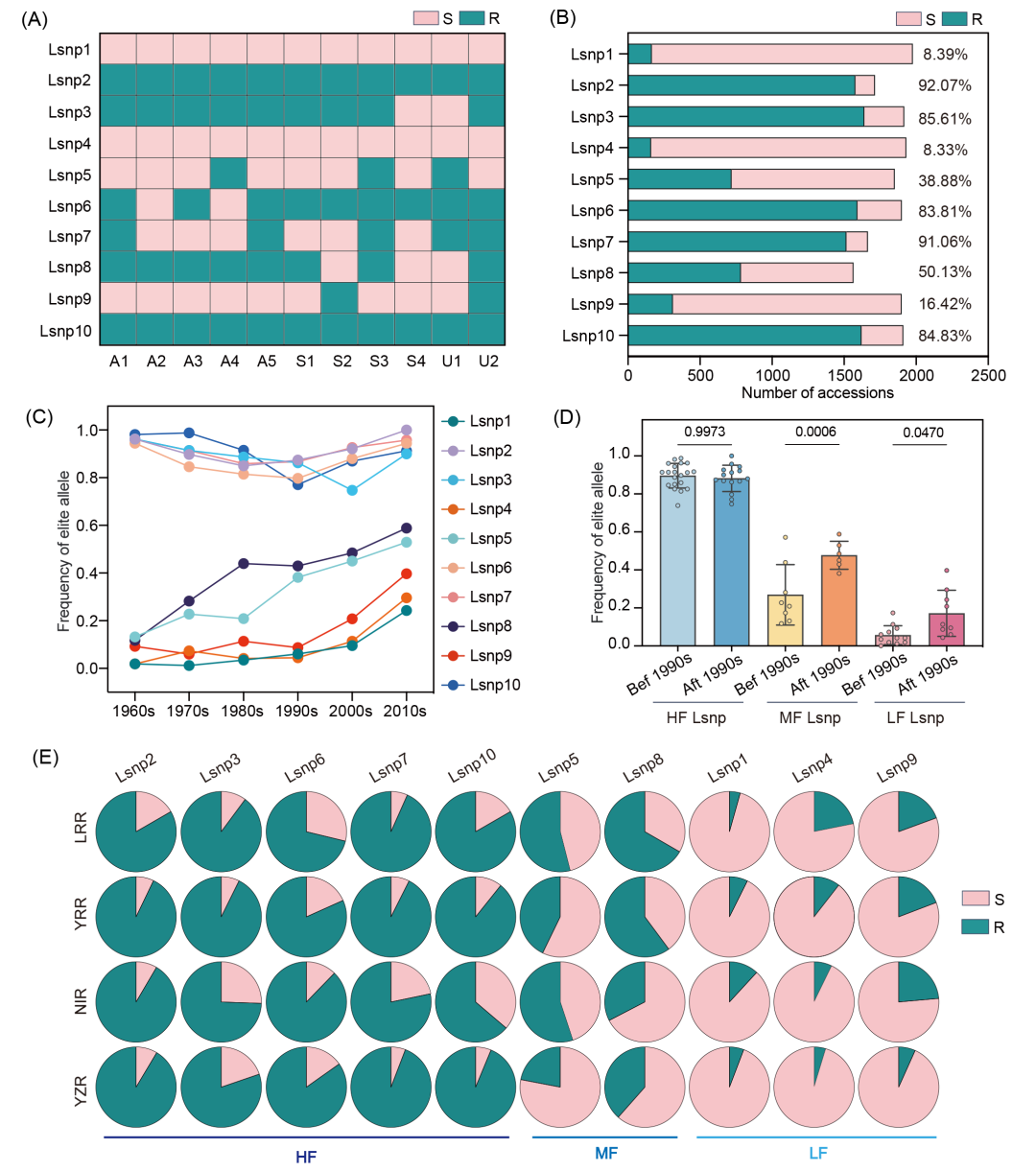
图2. 10个Lsnp抗性等位基因(LsnpR)的起源、分布与演化
(A) 基于国外引种祖先种质的LsnpR溯源分析。A1−A5代表美国祖先,S1−S3代表苏联祖先,U1−U2代表乌干达祖先。绿色表示LsnpR,粉色表示LsnpS;(B) LsnpR在中国棉花栽培品种(CDCs)中的频率分布。右侧数字表示携带LsnpR的品种占比;(C) 中国不同时期育成品种中LsnpR的利用频率;(D) 1990年代前后LsnpR利用频率差异(双尾t检验);(E) LsnpR在中国四大棉区的分布。HF/MF/LF分别表示高/中/低利用频率。
10个LsnpRs的聚合效应验证
为验证10个LsnpRs的聚合效应,本研究首先根据携带LsnpRs的数量将棉花品种分为10类来评估其聚合状态(图3A)。总体而言,71%−71.8%的棉花品种携带4−6个LsnpR(图S8A,B、表S4和S5),且携带LsnpRs的数量越多,棉花的抗病性越强(图3A)。值得注意的是,国家区试抗病对照品种中植棉2号(ZZM2)携带全部10个LsnpR,并在多环境中表现出稳定的抗病性(图S8C)。2033份CDCs中,仅有5份种质(冀资1号、冀资 96号、新陆中39号、新陆中40号和仁和39号)携带了10个LsnpR(表S4)。另外,基于公开发表的第三方自然群体的抗病鉴定数据,本研究进一步验证了10个LsnpR的聚合效应(图S8D,E)。
此外,针对10个LsnpR开发了棉花基因组病指预测模型。首先,采用岭回归模型估计各LsnpR效应值,构建了分子病指计算器(MDIC)。不同基因型组合的预测DI与观测DI显著相关(相关系数0.718)(图3B),表明该模型具有良好的预测准确性。尤为关键的是,低频率的Lsnp1R、Lsnp4R和Lsnp9R对黄萎病抗性具有最明显的提升效应,表明定向改良稀有位点可有效提升抗性(图S9)。
为进一步验证聚合效应和病指预测模型的准确性,本研究以ZZM2(抗病品种)与新陆早36号(XLZ36,感病品种)为亲本构建了包含272个家系的F2:3群体(图S10)。ZZM2携带10个LsnpR,而XLZ36仅携带Lsnp1R、Lsnp2R、Lsnp3R、Lsnp6R和Lsnp9R。通过测序确定了272个F2单株在Lsnp4、Lsnp5、Lsnp7、Lsnp8和Lsnp10位点的基因型(图S11),其中42、15、64、59和62个单株分别在Lsnp4R、Lsnp5R、Lsnp7R、Lsnp8R和Lsnp10R位点纯合(表S6)。选择在五个Lsnp位点均为纯合抗病基因型的19个F2单株(含13种组合模式)产生的F2:3家系进行接种验证(图3C和表S7)。结果显示,DI预测值越低的家系实际病指越低、维管束的病斑面积占比更小、病菌生物量更低,该结果表明聚合越多的LsnpR,棉花对黄萎病的抗性越强(图3D−G、S12和表S7)。
基于已发表的棉花抗黄萎病表型数据,本研究进一步验证了单个位点的抗性改良效果。以Lsnp1为例,选取仅Lsnp1基因型不同,其他Lsnp一致的品种系列(图3H),发现Lsnp1R品种系列的病指均显著低于Lsnp1S(图3H)。其余8个Lsnp(除Lsnp2因SNP检测未覆盖外)均获得类似验证结果(图3H−J、S13和表S8)。综上,本研究通过自然群体与人工群体多维度的验证,证实了聚合10个LsnpR可显著提升棉花对黄萎病的抗性。
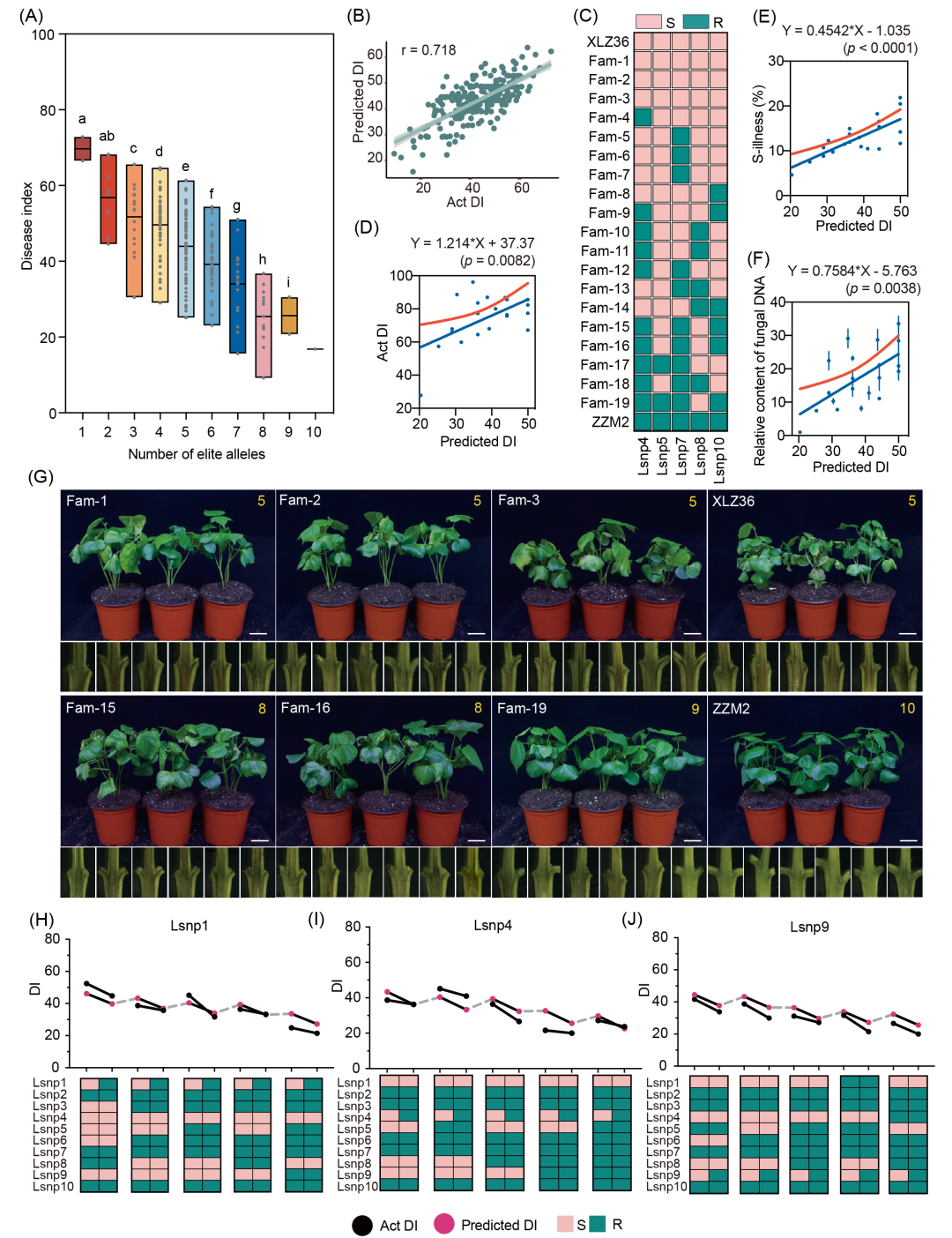
图3. 10个LsnpR的聚合效应
(A) 携带不同数量LsnpR材料的DI分布(All-b DI)。不同字母表示t检验差异显著(p < 0.001);(B) 分子病指计算器(MDIC)预测DI与实际DI的相关性;(C) 19个F2单株在5个Lsnp位点的基因型(ZZM2为抗病亲本,XLZ36为感病亲本);(D−F) F2:3家系实际DI、维管束病斑面积占比与病原菌生物量同预测DI的线性回归分析;(G) 基于预测DI筛选的极端抗/感F2:3家系表型。剖秆图为接种13天茎秆横切图。比例尺为3 cm;(H−J) LsnpR与LsnpS品种系列的基因型图谱及对比抗性提升效应。黑色点:实际DI;品红点:预测DI。
大丽轮枝菌胁迫响应基因主要参与ROS稳态与免疫应答调控
为解析棉花对大丽轮枝菌胁迫的转录响应规律,本研究对290份棉花群体材料进行对照与病原胁迫处理。根据前期研究发现的棉花-大丽轮枝菌互作阶段,分别于接种后3天(早期互作)和12天(晚期互作)采集根茎部样本,共获得4个处理组(正常条件M3、M12,胁迫条件D3、D12)的1152份转录组数据(图4A和S14)。主成分分析揭示病原侵染对基因表达具有广泛的影响,且早期与晚期侵染阶段呈现明显区分(图4B)。四组处理共鉴定59,317个表达基因(至少5%种质中FPKM > 0.1)(图4C)。基于20%以上种质表达量差异倍数 > 2的标准,筛选20,982个非冗余的胁迫响应基因(早期15,796个,晚期19,899个),占表达基因总数的35.37%(图S15A,B)。进一步根据表达模式将DEGs分为三类:病原诱导型(上调)、病原抑制型(下调)及响应变异型(图S15A,B)。
在生物胁迫响应中,叶绿体作为钙信号与活性氧(ROS)的信号源,通过将信号传递至细胞核激活防御基因表达。GO富集分析显示,病原诱导型基因主要参与转录调控及ADP结合(以NLR类蛋白为主);病原抑制型与响应变异型基因则显著富集于氧化还原过程、氧化应激响应、转录调控、系统获得抗性及蛋白磷酸化等通路(图S15C和S16),表明ROS稳态在棉花抗病中的核心作用。
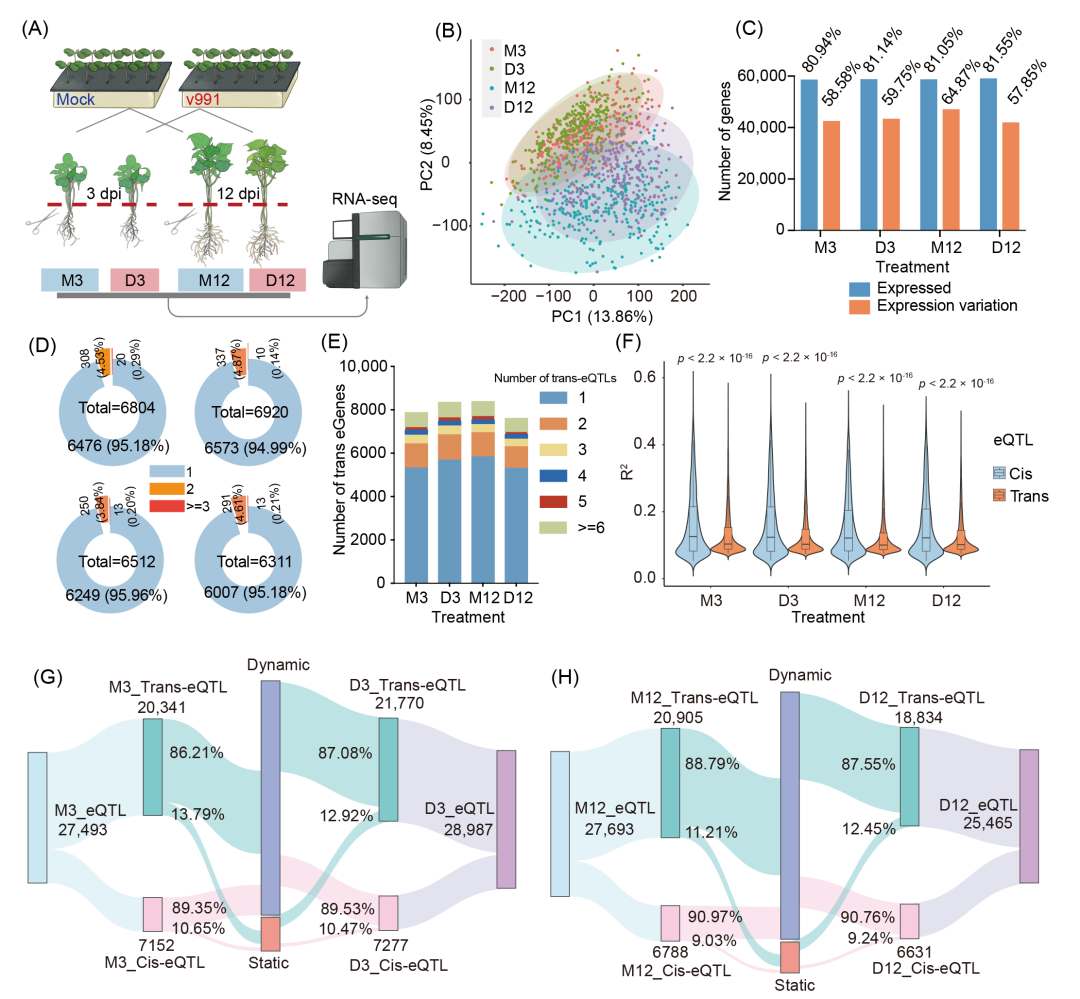
图4. 棉花黄萎病菌侵染早期与晚期基因表达与eQTL图谱
(A) 接种处理示意图。分别于接种后3天与12天取样进行RNA测序(M3/D3/M12/D12);(B) 1152个转录组样本的主成分分析;(C) 表达基因与差异表达基因数量统计;(D−E) 顺式eGene与反式eGene的eQTL数量分布;(F) 顺式与反式eQTL解释方差(R²)比较(双尾Wilcoxon秩和检验);(G−H) M3/D3与M12/D12处理间顺式与反式eQTL的静态/动态分布。
大丽轮枝菌侵染下基因表达的动态遗传调控解析
为探究遗传变异对棉花响应大丽轮枝菌基因表达的影响,保留48,430个表达量差异(5%−95%分位点范围超过2倍)的基因进行后续分析(图4C)。利用GWAS相同基因组变异数据集开展表达全基因组关联分析(eGWAS),共鉴定到27,848个顺式eQTL,调控10,771个基因(顺式eGene,即表达受顺式eQTL调控的基因)(图S17A−E和表S9)。其中仅约5%的基因受多个条件独立性eQTL调控,表明基因表达的遗传调控模式相对简单(图4D)。顺式eQTL显著富集于转录起始位点(TSS)附近,43.26%位于基因本体及其侧翼2 kb区域(图S17F,G)。同时鉴定到81,790个反式eQTL,调控21,420个基因(反式eGene)(图S17A−E和表S9),约30%的基因受多个反式eQTL调控(图4E)。值得注意的是,反式eQTL与靶基因间存在显著的At与Dt亚基因组间染色体互作偏好(图S17A−D),揭示了棉花抗病响应中复杂的染色体间遗传互作网络。
顺式与反式eQTL比较显示,顺式eQTL对表达变异的解释度(R²)在所有处理中均显著高于反式eQTL(图4F和S17A−D),表明顺式遗传变异对表达调控的影响更显著。多数eQTL在侵染早期与晚期呈现动态调控特征(图4G,H),提示大丽轮枝菌胁迫下的基因调控具有时序可塑性。进一步分析发现,41.19%的顺式eGene和75.25%的反式eGene与病原侵染相关(即特异性在侵染或对照条件下检出)(图S17H,I)。GO富集显示,这些eGene与胁迫响应基因功能相似,显著富集于氧化还原过程、光合作用及转录调控等通路(图S17J),证实基因组变异通过调控防御相关基因的表达影响抗病应答。
棉花抗大丽轮枝菌协同调控的基因模块与调控网络解析
为揭示多基因协同调控棉花抗大丽轮枝菌的机制,本研究通过独立成分分析(ICA)鉴定共表达基因模块。经分析,共识别到608个共表达模块(M3组152个、D3组158个、M12组142个、D12组156个),其中96个模块(M3组32个、D3组23个、M12组32个、D12组9个)与抗黄萎病显著关联(表S10)。进一步基于模块表达模式开展GWAS,发现各处理组均存在一个共表达模块(分别命名为M3-IC78、D3-IC7、M12-IC49、D12-IC36),其关联基因组位点与抗病QTL重叠:QTL1与四组模块均关联,QTL4特异性关联M12-IC49,QTL8关联除M12-IC49外的三组模块(图5A)。GO富集显示,四个模块中的基因均显著富集于ADP结合通路(图S18),涉及的基因主要编码NBS-LRR类抗病蛋白;M12处理组还富集ATP结合与蛋白磷酸化通路,涉及大量受体样蛋白激酶(RLK)基因(图S18),表明10个QTL中的QTL1、QTL4和QTL8可能通过调控经典抗病基因网络介导抗性。
eQTL热点区域指调控多基因表达的基因组聚集区。本研究共鉴定到306个热点区域,调控8249个基因(各热点调控25−286个基因),其中Dt亚基因组热点分布显著多于At亚基因组(Dt:At=197:109)。58个和46个热点分别特异性响应侵染早期与晚期阶段(图S19A和表S11),其调控基因主要参与ADP结合通路;13个热点与5个QTL(QTL1、QTL4、QTL5、QTL7和QTL9)存在共定位(表S12);超半数热点调控基因(156个)功能与病原响应基因相似,涉及氧化还原过程、细胞壁重塑及ADP结合等通路(表S13),该结果表明反式eQTL热点在胁迫下抗病基因表达调控中的核心作用。基于此,为了更直观地了解这些抗病相关的eQTL热点,本研究构建了包含这156个热点与4941个靶基因的棉花抗黄萎病基因调控网络(图S19B)。
棉花抗大丽轮枝菌相关基因的精细定位
为筛选抗黄萎病候选因果基因,本研究整合GWAS与eQTL数据,基于FUSION流程开展转录组关联分析(TWAS)。共鉴定47个独立的基因(M3组25个、D3组28个、M12组23个、D12组15个),其表达水平与病情指数(DI)显著关联(FDR校正p < 0.05)(图5A和S20A−D、表S14)。进一步采用孟德尔随机化(SMR)分析筛选一因多效性或因果关联导致表达水平与DI相关的基因,通过HEIDI检验(pHEIDI > 0.05)的25个基因中,19个与TWAS结果重叠,其中15个基因与5个GWAS定位的QTL重合(图5B,C),表明这些基因的顺式调控表达水平介导遗传变异与表型的关联。值得注意的是,QTL8鉴定到了最多的因果候选基因(6个),19个基因中13个受反式eQTL热点调控,其中位于QTL8区间内的GH_D07G1095是唯一受GWAS位点(QTL5)共定位热点(Hot102)调控的基因(表S12)。综上,我们整合GWAS与eQTL数据筛选出抗黄萎病QTL中15个高置信度候选因果基因(图5C)。
此外,TWAS Z值及基因表达与DI相关性分析显示,15个高置信度候选因果基因中有8个基因的表达水平与抗性呈负相关(图5C和表S16)。为验证其功能,本研究利用病毒诱导的基因沉默(VIGS)对8个负调控基因分别进行单基因沉默与多基因共沉默实验,结果表明8个负调控基因的单独沉默使棉花抗病性增强,共沉默8个基因对抗性提升效果最强,具体表现为棉花的黄化症状减轻、病指降低、维管束褐变减少及病原菌生物量下降(图5D,E和S21),该结果表明沉默负调控基因可有效提升棉花抗病性。
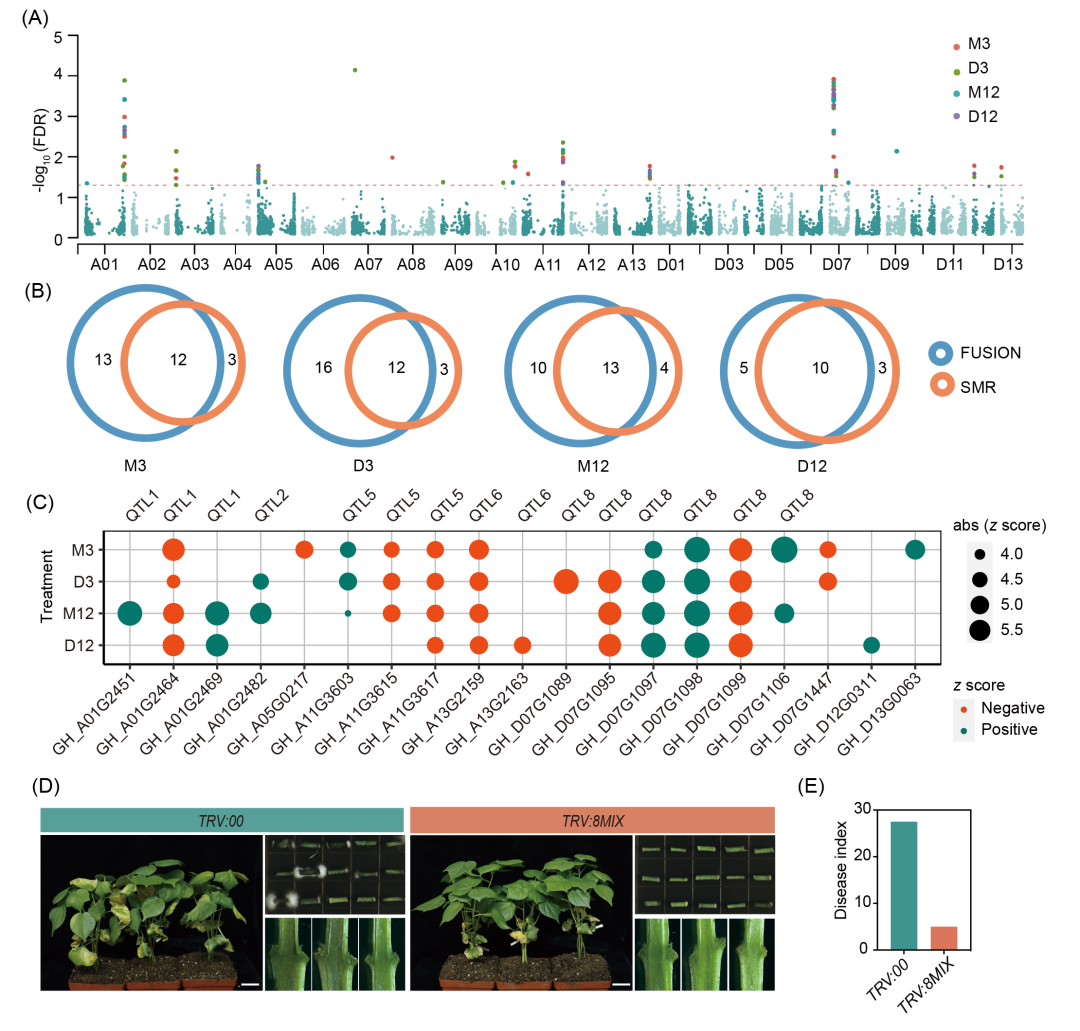
图5. GWAS与eQTL整合筛选棉花抗黄萎病候选基因
(A) 四组处理TWAS曼哈顿图。红色虚线为FDR校正阈值(−log10 (FDR) = 0.05;(B) FUSION与SMR方法筛选基因韦恩图;(C) 候选基因表型效应(TWAS Z值)气泡图。红点:负效应;深绿点:正效应;(D) TRV:00(对照)与TRV:8MIX(8基因共沉默)植株发病表型、病原菌恢复培养与维管束变褐情况。比例尺为3 cm;(E) TRV:00与TRV:8MIX植株DI统计。
GhARM通过调控ROS稳态介导棉花抗黄萎病机制
活性氧(ROS)稳态在棉花-大丽轮枝菌互作中发挥关键作用。在8个负调控抗性的基因中,QTL8内的GhARM(GH_D07G1095)编码含四个犰狳重复(ARM)结构域的未知功能蛋白。亚细胞定位分析显示GhARM定位于质膜、细胞质和叶绿体(图6A)。为验证该基因的功能,本研究通过CRISPR-Cas9系统构建了两个GhARM的敲除株系(GhARM-KO#1和GhARM-KO#2)(图6B)。实验表明,接种黄萎病菌后12天,与对照相比,敲除株系的叶片病症更轻、病指更低、维管束褐化更少(图6C−E)。真菌恢复培养实验证实突变体茎秆中病原菌量显著下降(图6F)。田间病圃鉴定显示,GhARM敲除株系对比野生型Jin668,在株高、果枝始节长、叶枝数和衣分等农艺性状无显著差异的情况下,抗性显著增强(图6G,H和S22)。
转录组分析发现,GhARM突变体中叶绿素生物合成通路显著富集,相关基因表达上调(表S17和图S23)。叶绿素含量测定显示突变体叶绿素a/b含量高于野生型(图6I)。差异基因同时富集于氧化还原过程、氧化应激响应及植物免疫通路,其中过氧化物酶基因表达显著下调(表S17和图S23)。为验证GhARM通过调控ROS稳态影响抗性,本研究检测了flg22与几丁质诱导的ROS爆发情况,结果表明突变体中ROS积累显著增加(图6J,K)。利用H2DCFDA探针检测叶绿体ROS(cROS),突变体中的绿色荧光信号显著增强(图6L,M),表明GhARM可能通过调控cROS稳态介导抗性。
基于290份种质的GhARM自然变异分析鉴定出HAP1和HAP2两种主要单倍型。携带HAP2的种质基因表达量更低且病指更低,表明GhARMHAP2具有更强的抗性(图S24A,B和S25)。进一步对3516份陆地棉种质(含237份野生型、138份地方品种和3141份栽培种)的GhARM上游2 kb及编码区进行变异分析,共检测10个SNP和2个Indel,形成7种主要单倍型(HAP1−HAP7)(图S24C,D)。野生型中单倍型多样性较高(以HAP2和HAP3为主),地方品种中HAP2频率上升,而现代栽培种中HAP2比例未显著变化(图S24E,F),显示出该基因具有改良潜力。GhARM侧翼300 kb区域的核苷酸多样性(π)分析显示,野生型多样性高于地方品种与栽培种(图S24G);Tajima's D值分析表明,栽培种与地方品种多呈负值,而野生型多为正值(图S24H),反映该区域在驯化过程中受人工选择压力。综上,GhARM在棉花驯化与改良中经历人工选择,可为抗病育种提供新靶点。
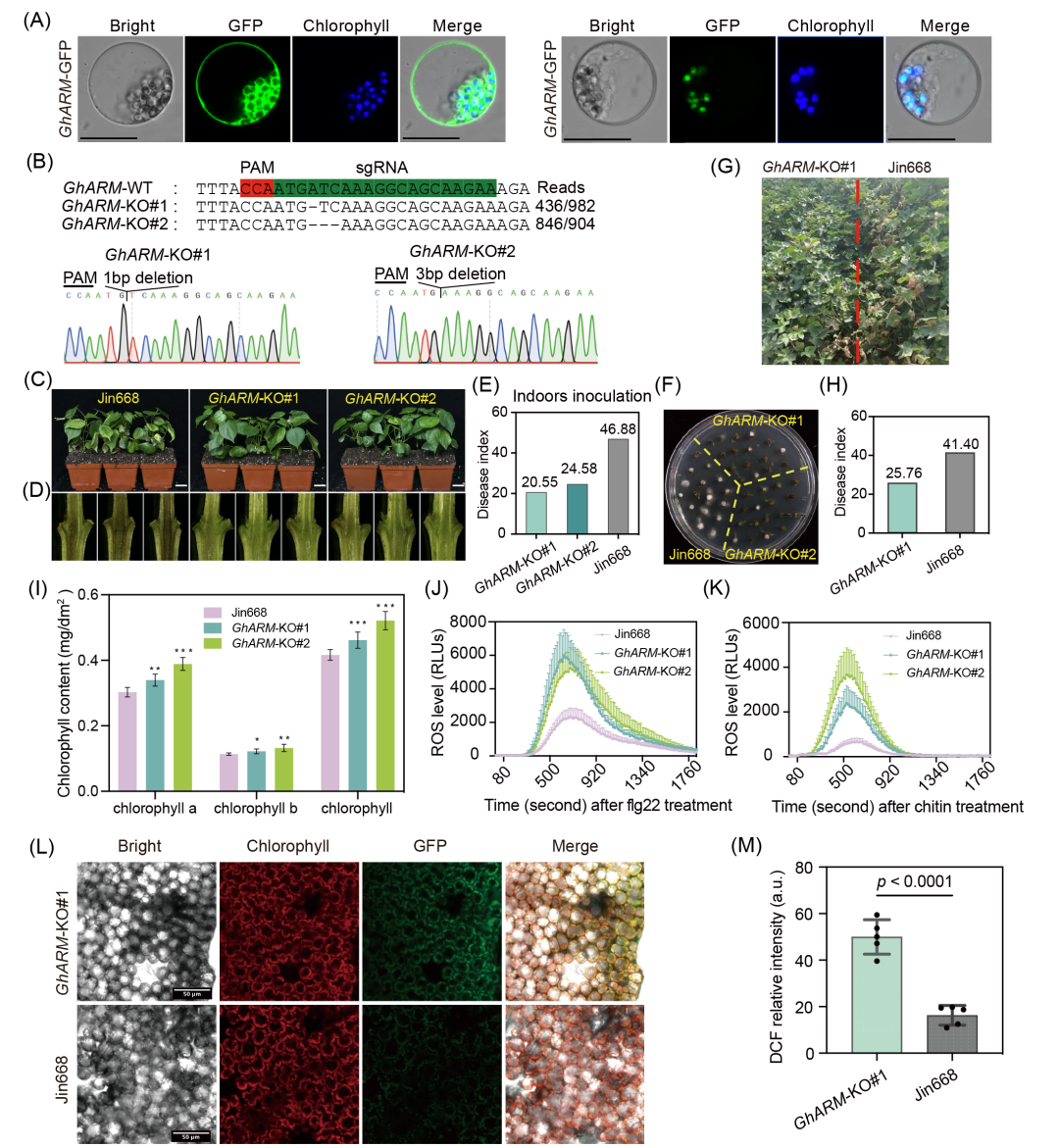
图6. GhARM通过调控ROS爆发介导棉花抗黄萎病
(A) GhARM在棉花原生质体中的亚细胞定位,比例尺20 μm;(B) GhARM敲除株系突变类型(Jin668背景);(C−F) 野生型与敲除株系接种v991后12天的发病表型(C)、维管束染色(D)、DI(E)与真菌恢复培养(F)。比例尺为3 cm;(G−H) 田间条件下Jin668与GhARM敲除株系抗性比较及DI统计;(I) 叶绿素a/b及总量测定(单因素ANOVA,*p < 0.05,**p < 0.01,***p < 0.001);(J−K) flg22(J)与几丁质(K)诱导的ROS爆发检测;(L−M) 共聚焦显微镜观察接种3天后叶肉细胞H₂O₂积累(H2DCFDA染色)及荧光强度定量(t检验,n = 5)。
讨 论
优异等位基因挖掘与聚合协同提升棉花黄萎病抗性
秸秆还田与连作制度导致病原菌在病圃土壤中持续积累,加之大丽轮枝菌菌系组成复杂、土壤微生物群落互作以及陆地棉抗源匮乏,共同加剧了棉花黄萎病防控的复杂性。尽管已报道了200多个抗黄萎病QTL及微效基因,但QTL定位精度不足、功能基因机制研究欠深入,制约了抗病遗传标记与基因在抗病育种中的有效应用。类似于油菜的含油量、棉花的纤维,聚合育种是改良棉花抗黄萎病等复杂性状的高效手段。研究发现,在新疆黄萎病病圃中分离的病原菌在生长特性与致病力上存在显著差异,在薄荷、橄榄和马铃薯黄萎病的病圃有一致结论。本研究鉴定的10个QTL在多环境中表现稳定效应,其中qVWR.A10.3(QTL4)和qVWR.D07.1(QTL8)与已报道QTL存在重叠,其余8个QTL均为首次发现。无论是田间自然发病还是人工接种,聚合全部10个LsnpR均能显著提升抗性,中植棉2号(ZZM2)携带全部LsnpR为其稳定抗性提供了遗传解释。
值得注意的是,菌系的地理特异性、不同的耕作方式会显著影响棉花的抗性表现,进而决定品种被选择的命运。我国育种中固定利用的Lsnp1R和Lsnp4R是在病圃压力下筛选的优异位点,具有最显著的抗性效应与稳定性。类似地,陆地棉抗枯萎病育种经历了连续多年在重病地进行单株选育,在强大的选择压力下,主效抗病基因Fov7的单核苷酸变异为枯萎病育种带来了曙光。自20世纪90年代以来,Lsnp1R、Lsnp4R、Lsnp5R、Lsnp8R和Lsnp9R的利用频率显著增加,可能与90年代至21世纪初黄萎病大规模爆发后,以抗病育种作为主要目标的历史时期密切相关。我们推测,中、低频的LsnpR在育种进程中对抗性提升具有重要价值。不同年代品种中10个LsnpR的频率动态变化呈现显著波动性而非持续线性增长,暗示部分LsnpR可能尚未被纳入系统的定向选择体系,这可能归因于抗病与产量性状的权衡——产量改良可能以抗性降低为代价。未来抗病育种中,可借助分子病指计算器(MDIC)预测基因型抗性水平,针对性导入LsnpR(尤其是Lsnp1R、Lsnp4R、Lsnp5R、Lsnp8R和Lsnp9R),实现抗性精准提升。
陆地棉单一的遗传背景是限制抗病性改良的瓶颈。Fhb7,一个来源于小麦近缘野生种长穗偃麦草(Th. elongatum)的基因,赋予小麦对赤霉病的持久抗性。在中国棉花育种史上,育种家也采用了远缘杂交的手段尝试为陆地棉带来产量、品质特别是抗性方面的提升。研究表明,亚洲棉(G. arboretum)基因组中存在着抗黄萎病的基因资源。本研究中ZZM2的育种过程也经历了与抗黄萎病的海岛棉(G. barbadense)品种"多毛早"的杂交,这表明通过挖掘和利用其他棉种(系)的抗病基因资源库,是突破陆地棉遗传背景同质化限制的有效策略。
表达数量性状基因座(eQTL)在生物胁迫下防御相关基因表达多样性的形成中发挥重要作用
全基因组关联分析(GWAS)已被证明是研究多种作物复杂性状遗传基础的有效方法。在后GWAS时代,整合GWAS与eQTL分析通过调控靶基因表达来解析复杂性状的遗传变异机制,已成为深入理解其遗传基础的重要策略。尽管已有研究关注植物在非生物胁迫(如干旱胁迫和热胁迫)下调控基因表达的遗传变异,但据我们所知,本研究首次揭示了生物胁迫下调控基因表达的遗传变异。本研究发现109,638个eQTL与25,187个基因相关联,为棉花抗黄萎病的后GWAS研究提供了宝贵资源。大丽轮枝菌胁迫下的差异表达基因(DEGs)与eQTL调控基因具有相似的功能特征,表明基因组多样性对抗病相关基因表达具有重要影响。这可能与中国棉花育种历史相关,黄萎病长期作为棉花生产的主要威胁,育种家长期致力于选育抗黄萎病品种,导致不同棉花品种的基因组多样性与抗病基因表达形成密切关联。通过整合GWAS和eQTL数据,本研究阐明了棉花抗黄萎病的遗传基础和调控机制,不仅为棉花抗病研究提供新见解,也为植物响应病害胁迫的机制研究提供了更广泛的启示。
活性氧稳态在防御信号中发挥重要作用
随着研究的深入,越来越多的作物中发现了具有负调控作用的感病基因有很大的利用价值,如小麦的Mlo、水稻的SWEET、小麦中编码RLCK的 TaPsIPK1等。通过基因编辑技术来编辑负调控基因也成为了一种经济、高效的策略来提高作物的抗性。陆地棉遗传背景狭窄、抗病种质资源匮乏且整体抗病性较差,导致抗病基因效应微弱且难以有效应用于育种实践。因此,相较于单纯依赖抗病基因,通过挖掘棉花易感基因并利用基因组编辑技术进行靶向编辑,是培育抗黄萎病品种的有效策略。本研究验证了8个负调控基因的功能,同时沉默8个基因可进一步增强棉花对黄萎病菌的抗性,但这些负调控基因间潜在的协同调控效应及其作用机制仍需深入解析。通过基因编辑敲除其中一个负调控基因GhARM后,室内接种与田间抗病性鉴定均显示棉花对黄萎病的抗性显著增强,未来研究可采用多基因编辑系统同时靶向这些负调控基因并创制无Cas9植株。
GhARM基因定位于叶绿体,提示其可能通过调控叶绿体介导的活性氧(ROS)稳态负调控棉花对黄萎病的抗性。随着研究进展,作为光合作用与植物生长核心器官的叶绿体,其免疫调控功能日益受到关注。叶绿体是逆境条件下ROS产生的主要细胞器,但黄萎病菌侵染过程中叶绿体ROS稳态的维持机制尚不明确。本研究发现敲除GhARM后棉花植株在黄萎病菌胁迫下叶绿体ROS水平升高,这可能是其增强抗病性的重要机制。前期研究发现黄萎病菌通过诱导ROS产生促进侵染,提示叶绿体ROS与总ROS在棉花抗黄萎病过程中可能具有不同作用方向。由于叶绿体在植物生长与免疫调控中具有重要作用,其在植物-病原互作过程中易成为病原菌攻击靶标。GhARM如何调控叶绿体ROS稳态仍需进一步研究。
结 论
本研究系统解析了棉花抗黄萎病的遗传基础与调控网络,阐明了棉花抗黄萎病育种改良的遗传学机制,为复杂农艺性状中数量性状位点(QTL)聚合的作用提供了重要见解。通过多环境鉴定的10个QTL,我们构建了高效的棉花抗黄萎病基因组选择育种模型,并利用F2:3群体验证了该模型对棉花抗黄萎病表型的预测能力,为基于基因组选择的棉花抗病育种提供了新策略。整合基因组、群体转录组与功能基因组学分析,揭示了活性氧(ROS)在棉花抗黄萎病中的核心调控作用。研究成果为棉花抗病育种提供了宝贵的遗传资源,并为解析棉花及其他作物抗黄萎病的遗传基础和调控机制提供了重要理论依据。
方 法
植物材料与田间试验
本研究使用的290份陆地棉种质资源来自本实验室完成重测序的自然群体。材料种植于新疆玛纳斯(M)、库尔勒(K)和库车(C)的自然黄萎病圃。分别在病害发生期采集了1年(M)、2年(K)和2年(C)的病情指数(DI),包括5个独立环境(18M、18K、18C、19K、20C)。除20C为单次重复外,其余年份和地点均设置两次重复,共获得9组原始DI数据(18M-1、18M-2、18K-1、18K-2、19K-1、19K-2、18C-1、18C-2、20C-1)。基于最佳线性无偏估计(BLUE)对数据进行整合,生成M-b、18K-b、18C-b、19K-b、K-b、C-b共6组DI数据。进一步对9组原始DI进行BLUE分析,获得综合DI数据(All-b)。此外,筛选各年份/地点中DI较高的重复样本,生成18M-3、18K-3、18C-3、19K-3共4组合并DI数据,最终形成20组DI数据集(图1A)。
基因组变异检测
基于前期研究产生的重测序数据,使用fastp(v0.20.0)进行质控,采用Sentieon(v201911)标准流程进行SNP检测。具体流程为:通过BWA(v0.7.17)将质控后reads比对至TM-1参考基因组;利用SAMtools(v1.9)去除重复与低质量比对序列;使用Sentieon Haplotyper算法(参数:--algo Haplotyper --emit_conf=10 --call_conf=10 --emit_mode gvcf)生成单样本GVCF文件;通过GVCFtyper算法合并所有样本变异信息,获得整合VCF文件;最终利用VCFtools(v0.1.16)过滤保留深度(DP)> 5、缺失率(missing)< 50%、最小等位基因频率(MAF)> 0.05的高质量SNP。
全基因组关联分析(GWAS)
基于290份群体中提取2,152,153个高质量SNP。采用GEMMA(v0.98.1)混合线性模型进行20组DI表型与基因变异的关联分析。基于STRUCTURE(v2.3.4)对10万个SNP子集进行群体结构分析,利用全基因组SNP通过GEMMA计算亲缘关系矩阵。GWAS显著性阈值设定为经验阈值1E-05和Bonferroni校正阈值4.65E-05(1/n,n为总SNP数)。将同一连锁不平衡区块(LD,r² ≥ 0.5)内的显著位点合并为单个QTL。
基于岭回归的DI表型基因组预测
使用glmnet R包(v4.0-2)通过岭回归建立预测模型。以10个稳定QTL为预测变量,20组DI为响应变量,通过cv.glmnet()函数(alpha=0)进行模型拟合。采用交叉验证选择最优超参数λ(正则化系数),以最小化交叉验证均方误差(MSE)。最终通过predict()函数实现表型预测。
群体转录组测序
在华中农业大学温室采用水培法种植290份材料(图4A)。将材料固定于42 cm×30 cm塑料托盘,使用霍格兰营养液(NS10205,Coolaber,北京)培养,每份材料准备10株幼苗。以v991菌株接种(Czapek培养基,MM1020-C,Coolaber,北京),接种浓度为1×10⁷ spore/mL,设置对照处理(同浓度Czapek溶液)。播种15天后分别进行接种与对照处理,于处理后3天和12天采集样本(除J17、J36外,每处理组含288份材料),共获得1152个样本。使用RNAprep Pure Plant Plus Kit(DP441,天根,北京)提取RNA,于派森诺生物科技有限公司(上海)构建文库(VAHTS Universal V6 RNA-seq Library Prep Kit,NRM604-02,诺唯赞,南京)并在NovaSeq 6000平台进行双端150 bp测序。
eQTL定位分析
将表达量FPKM值 > 0.1且超过5%材料表达的基因定义为表达基因,进一步过滤表达变异(5th与95th百分位间差异 < 2倍的基因)。经筛选后,四组处理(M3、D3、M12、D12)分别保留42,173、43,014、46,703和41,644个基因进行eQTL分析。为消除批次效应和混杂因素,采用PEER(v1.0)方法估算潜在混杂因子。将估算因子作为协变量以提高分析效能。利用Plink基于基因型数据计算主成分(PCA)以评估群体结构。通过R包"qqnorm"对基因FPKM进行分位数标准化,并采用TensorQTL(v1.0.8)的线性回归模型进行eQTL定位,以前20个PEER因子和前5个PCA作为协变量。
将位于基因转录起始位点上下游1 Mb内的eQTL定义为顺式eQTL(cis-eQTL),其余则归为反式eQTL(trans-eQTL)。对顺式eQTL,通过置换检验与逐步回归法筛选条件独立性位点,参数设置为--cis --window 1000000 --qvalue_lambda 0.05。该方法基于Beta分布计算经验p值并校正全基因组错误发现率(FDR),q值<0.05的SNP被定义为显著顺式eQTL,对应基因为顺式调控基因(cis-eGene)。
对于反式eQTL,首先去除基因1 Mb内的顺式关联信号,采用GEC(v1.0)计算独立性SNP有效数量以确定显著性阈值,建议p值为2.45E-06(1/364,248)。由于反式eQTL数量庞大,对每个基因连续10 kb区域内 ≥ 3个显著SNP的位点进行聚类,选取最显著SNP作为代表性eQTL。同时合并同一连锁不平衡区块(R² > 0.1)内的eQTL。通过置换检验(1000次)结合滑动窗口(1 Mb窗口,100 kb步长)识别反式eQTL热点区域,显著性阈值设定为p = 0.01。
棉花抗黄萎病TWAS分析
基于FUSION软件包,以顺式调控基因表达量为表型,1 Mb内SNP为基因型,构建PLINK格式文件。通过FUSION compute_weights.R脚本(使用BLUP、BSLMM、LASSO、Elastic Net和top SNPs五种模型)计算基因表达权重。利用FUSION.assoc_test.R整合GWAS结果与表达权重进行关联分析,FDR校正p值 < 0.05的基因被判定为显著关联基因。
SMR共定位分析
使用SMR(v1.03)软件的SMR & HEIDI方法评估SNP对黄萎病抗性效应是否受基因表达调控。将各处理组顺式eQTL数据转换为BESD格式,整合GWAS结果(20组DI)进行SMR分析。以p值 < 1/n(n为顺式eGene总数)作为显著性阈值。
ICA共表达模块构建
通过R包"picaplot"(v0.99.7)对标准化表达矩阵进行独立成分分析(ICA),参数设置为var_cutoff=80、max_iter=10、n_runs=15(https://github.com/jinhyunju/picaplot)。分解矩阵X = S × A,其中S矩阵(基因×成分)用于基因模块划分,A矩阵(成分×样本)表征成分表达模式。利用EMMAX(v20120205)分析各成分与黄萎病抗性关联。
载体构建
VIGS载体:针对8个候选基因设计300-500 bp特异片段,克隆至TRV载体的BamHI/KpnI位点(ClonExpress II一步克隆试剂盒,C112,诺唯赞,南京)。
CRISPR载体:将靶向GhARM的sgRNA克隆至pRGEB32载体(GhU6启动子驱动)。
瞬时表达载体:扩增GhARM编码序列并克隆至pHBT载体(CaMV 35S启动子驱动,C端融合GFP标签)。引物信息见表S18。
病原菌接种实验
采用黄萎病菌落叶型菌株v991接种棉花。菌株经PDA平板活化后转接至Czapek-Dox液体培养基(25℃摇培3−4天),纱布过滤后调整孢子浓度至2×10⁵ spore/mL。通过根浸法接种(浸根2分钟),移栽至营养土-蛭石混合基质。接种12天后观察发病情况并计算病情指数。
对于Jin668和GhARM突变体材料,接种v991后分别于0和3天采集根部样本进行RNA测序。
叶绿素含量测定
取各基因型叶片打取6片叶盘(直径5 mm),浸于1 mL 95%乙醇(4℃避光浸提12小时)。使用EnSpire多功能酶标仪(PerkinElmer)测定645 nm和663 nm吸光值,按公式计算含量:
叶绿素a(μg/mL)= (12.72A663 - 2.59A645) × V/W × 1000
叶绿素b(μg/mL)= (22.88A645 - 4.67A663) × V/W × 1000
总叶绿素(μg/mL)= (20.29A645 + 8.05A663) × V/W × 1000
(V:提取液体积/mL,W:叶面积/dm²)。
ROS检测
化学发光法:取野生型与GhARM突变体三周龄叶片制备5 mm叶盘,预浸无菌水(室温过夜)后,加入50 μM鲁米诺(Sigma-Aldrich)+20 μg/mL辣根过氧化物酶(Sigma-Aldrich),分别添加100 nM flg22或100 μg/mL几丁质诱导。通过EnSpire酶标仪记录30分钟内化学发光信号。
荧光探针法:参照文献方法,叶盘经25 μM H2DCFDA(避光孵育30分钟)染色后,使用Leica SP8共聚焦显微镜观察(激发488 nm,发射500-600 nm;叶绿素自发荧光检测640-735 nm)。
代码和数据可用性
本研究所使用的原始测序数据已存储在美国国家生物技术信息中心(NCBI)数据库,RNA测序数据的项目登录号为PRJNA1195373(访问链接:https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1195373)。主要数据和代码已上传至Github网站,网址为:https://github.com/smliu-lshm/cotton_VW_QTL。更详细的数据信息可联系通讯作者获取。补充材料(文本、图、表、中文翻译版本或视频)也可从线上(http://www.imeta.science/)获取。
引文格式:
Xiaojun Zhang, Shiming Liu, Peng Wu, Wanying Xu, Dingyi Yang, Yuqing Ming, Shenghua Xiao, et al. 2025. “A panoramic view of cotton resistance to Verticillium dahliae: from genetic architectures to precision genomic selection.” iMeta 4: 70029. https://doi.org/10.1002/imt2.70029.
作者简介

张晓君(第一作者)
● 华中农业大学作物遗传育种专业在读博士研究生。
● 研究方向为基于多组学解析棉花抗黄萎病遗传基础与调控机制,以第一作者在iMeta发表论文1篇,相关研究成果申请国家发明专利2项,国际专利1项。

柳仕明(第一作者)
● 华中农业大学植物科学技术学院博士后。
● 研究方向为棉花与枯、黄萎病菌的互作,以第一作者或通讯作者(含共同)在iMeta、Advanced Science、Journal of Fungi等期刊发表学术论文3篇。博士后工作期间,主持国家自然科学基金青年项目、中国博士后科学基金面上项目、中国博士后科学基金特别资助以及湖北省博士后创新人才项目。

朱龙付(通讯作者)
● 华中农业大学教授,博士生导师。作物遗传改良全国重点实验室副主任,挂任黑龙江省农业科学院党组成员、副院长,兼任中国农学会棉花分会副主任、湖北省棉麻学会理事长,国家棉花生物育种科技创新联盟秘书长。2022年入选国际棉花基因组委员会(ICGI)功能基因组共同主席。
● 主要研究方向为棉花与病原分子互作及早熟多抗分子育种,鉴定到陆地棉抗枯萎病主效基因Fov7和调控棉花抗黄萎病关键基因GhSSN等,在Advanced Science、Nature Communications等国际学术期刊发表研究论文100多篇,其中3篇论文入选Web of Science高被引/热点论文,申请或获得国家发明专利20多项。相关研究成果获得国家科技进步二等奖一项(2013)、教育部、湖北省和新疆自治区科技进步奖一等奖等6项。担当JIPB等国际学术期刊编委,2018年入选国家级人才项目计划。

张献龙(通讯作者)
● 华中农业大学教授,博士生导师。中国工程院院士,首批万人计划领军人才,首批全国创新争先奖,全国先进工作者,新中国培养的第一个棉花领域农学博士,国家教学名师,中国作物学会棉花专业委员会会长、国际棉花基因组委员会(ICGI)联合主席/主席(2021−2023/2023−2025)。
● 在Nat Genet、Nat Plants等学术期刊发表研究论文300余篇。培育出华杂棉H318等25个棉花新品种,其中国审品种6个。爱思唯尔“中国高被引学者",2020年以来连续入选科睿唯安“全球高被引科学家”。

金双侠(通讯作者)
● 华中农业大学教授、博士生导师。现任植物科学技术学院院长,曾获得国家杰出青年基金获得者、教育部青年长江学者、新疆天池英才等人才项目。
● 目前主要从事棉花生物技术、基因编辑;棉花与害虫互作分子机制等研究工作。近年来开发了CRISPR/Cas9,Cas12a,Cas12b,CBE/ABE/CABE/ABE8e,Cas13a/b/c(转录抑制), 转录激活,m6A甲基化/去甲基化表观修饰等10余套基因编辑工具,在国际主流学术期刊Nature Genetics、Nature Communications、Genome Biology、Advanced Science、iMeta、Trends in Plant Science、Plant Biotechnology Journal等杂志上发表 130 多篇植物生物技术相关论文,其中12篇论文入选Web of Science高被引/热点论文,论文引用8200多次。目前担任Plant Biotechnology Journal杂志(影响因:11.2)执行主编、Genome Biology(影响因子10.1)编辑、iMeta副主编(影响因子:23.8)、Crop Journal(影响因子:6.0)副主编;2017−2021年担任棉花基础研究领域权威学术机构-国际棉花基因组协会(ICGI)共同主席。2021年获得棉花基础研究领域权威奖项-Cotton Biotechnology Award。
更多推荐
(▼ 点击跳转)
iMeta | 引用16000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期
iMetaOmics封面

1卷1期

1卷2期

2卷1期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2024年6月获得首个影响因子23.8,中科院分区生物学1区Top,位列全球SCI期刊前千分之五(107/21848),微生物学科2/161,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆11/514!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,是定位IF>10的高水平综合期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
邮箱:
office@imeta.science

















 1272
1272

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









