






点击蓝字 关注我们
食叶猴肠道微生物组的物种水平探索反映了人类活动的影响和特化的饮食生态位:论第四层级生物多样性的保护

iMetaOmics主页:http://www.imeta.science/imetaomics/
研究论文
● 期刊:iMetaOmics
● 原文链接: https://onlinelibrary.wiley.com/doi/10.1002/imo2.70051
● DOI: https://doi.org/10.1002/imo2.70051
● 2025年9月16日,广西师范大学大学周岐海、郭秋艳和南京中医药大学朱立峰、崔新远等在iMetaOmics在线发表了题为“Species-level exploration of the gut microbiome in the leaf-eating Presbytis monkeys reflected the effects of anthropogenic activity and specialized dietary niches: conservation on the fourth biodiversity level”的文章。
● 本研究通过宏基因组分析发现,人类干扰力度越大,则叶猴肠道中人类相关微生物比例会越高,这些也可能与饮食变化有关。研究揭示了肠道微生物协助宿主降解纤维素与草酸盐的适应机制,并提出“共生微生物多样性”作为生物多样性保护第四层次。
● 第一作者:周岐海、郭秋艳、崔新远、孟涛
● 通讯作者:朱立峰(zhulf2020@126.com)、周岐海(zhouqh@ioz.ac.cn)
● 合作作者:庞竹婷、 王松、陈华、卢美静、刘国琦、黄乘明
● 主要单位:广西师范大学、南京中医药大学、广西林业勘测设计院、广西南宁动物园、明科生物科技有限公司、中国科学院动物研究所
亮 点
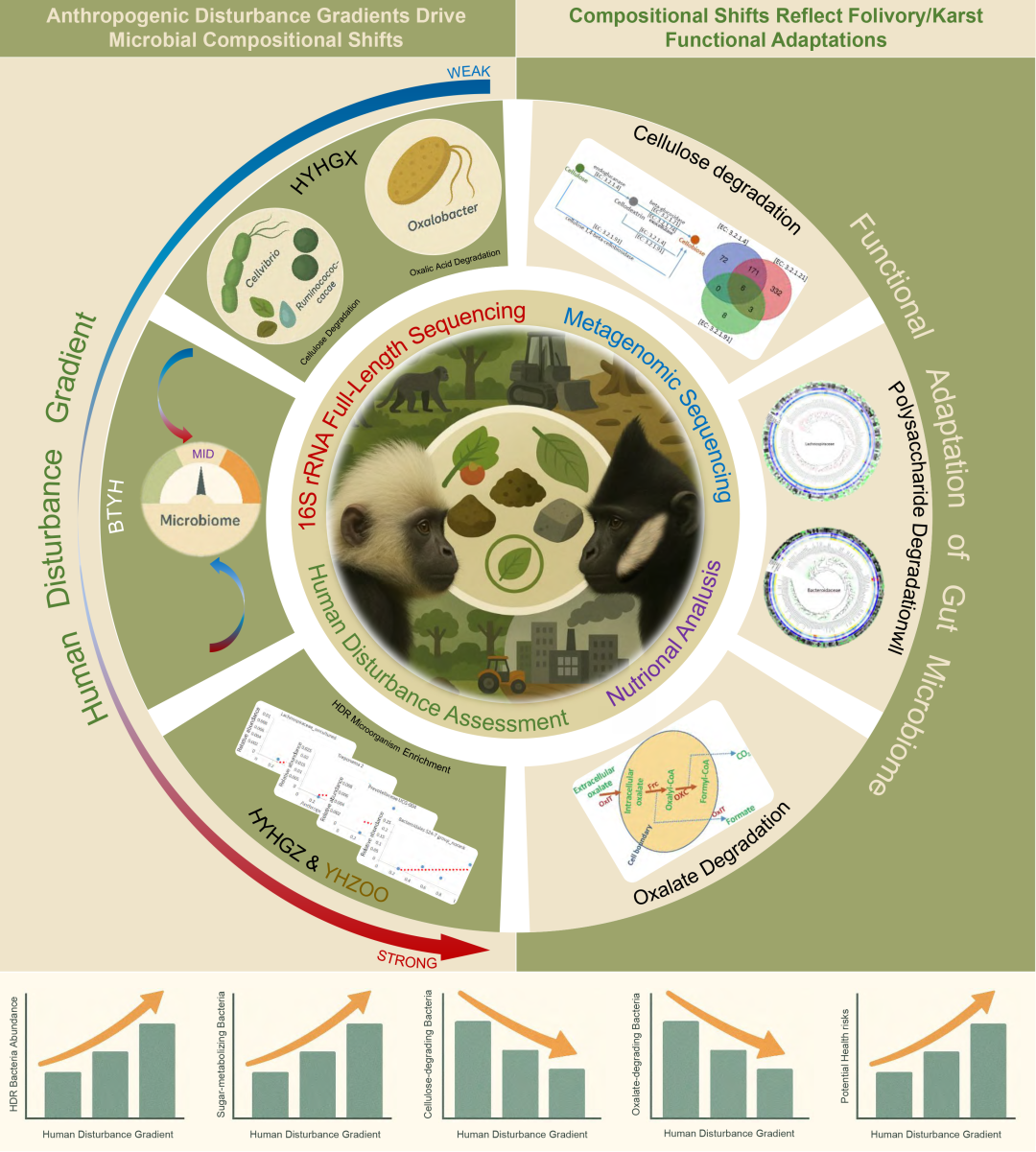
● 对濒危Presbytis叶猴的物种水平肠道微生物组分析揭示了从野生到圈养环境的人为干扰梯度上存在明显的微生物转变;
● 基于1,199个高质量MAGs的功能宏基因组学揭示了微生物在纤维素和草酸盐降解方面的适应,支持宿主在高纤维、高草酸盐的石灰岩森林饮食中生存;
● 人类相关类群(如毛螺菌科、普雷沃菌科、密螺旋体属)丰度的增加与接触人类食物和环境相关,表明微生物组的“人源化”;
● 本研究提出“共生微生物多样性”作为遗传多样性、物种多样性和生态系统多样性之外的生物多样性第四层级,强调了其保护价值。
摘 要
全球范围内,75%的非人灵长类动物种群数量下降,其主要原因是由人类干扰导致的森林砍伐和栖息地丧失。濒危的白头叶猴和黑叶猴是两种栖息于石灰岩森林的异域分布物种。鉴于人类干扰对肠道微生物群落的负面影响,我们采用多种方法(景观遗传学、营养分析、行为观察、237个样本的全长16S rRNA基因测序以及深度宏基因组测序),研究了这两种野生叶猴在人类干扰下(以动物园作为最强干扰的参考)的肠道微生物组成和功能。我们发现,四种典型的人类干扰相关(Human Disturbance-Related, HDR)肠道微生物科(毛螺菌科 Lachnospiraceae 和拟杆菌科 Bacteroidaceae 等)的相对丰度随着人类干扰的增强而增加。基于对1,199个非冗余宏基因组组装基因组(MAGs)的比较基因组学分析,我们鉴定出了参与主要膳食或宿主来源成分(如,膳食来源的多糖、草酸盐和宿主来源的聚糖)消化的肠道微生物物种。这些叶猴携带了一些推测共有的纤维素分解菌和草酸盐降解菌,同时也携带了一些宿主特异性的细菌。因此,我们得出结论,叶猴种群中HDR物种的增加可能是由饮食变化和糖摄入量增加引起的,例如动物园中的玉米和高粱馒头、水果,以及野生叶猴(特别是HYHGZ种群)中的人类食物、玉米和红薯。在人为干扰下,饮食、宿主和生存环境等因素可能影响这些叶猴的肠道微生物群落和功能。叶猴肠道微生物组在帮助宿主适应叶片饮食和石灰岩生存环境方面发挥着潜在作用。未来,野生灵长类的保护不仅应关注宿主遗传多样性,还应关注人为干扰下共生微生物组的变化。
视频解读
Bilibili:https://www.bilibili.com/video/BV1sVHGzUEd1/
Youtube:https://youtu.be/dLBDzBBkGfU
中文翻译、PPT、中/英文视频解读等扩展资料下载
请访问期刊官网:http://www.imeta.science/
全文解读
引 言
生物多样性通常在三个层次上进行探索:遗传多样性、物种多样性和生态系统多样性。野生动物濒危的主要原因是人类活动导致的栖息地丧失和碎片化。野生动物保护主要关注这三个层次。例如,一些研究涉及野生动物的遗传评估。遗传多样性是哺乳动物进化潜力的指标 。然而,大约100万亿微生物栖息在人体系统中,其中约95%存在于肠道系统。肠道微生物组在宿主营养摄入、发育和健康中扮演重要角色。肠道微生物组也有助于野生哺乳动物增强其本地适应性,因此在宿主生存和保护中起着至关重要的作用。
近来,人类开发与破坏(如森林砍伐、侵占、城市化和食物投喂)已被证明对野生动物的肠道微生物组产生负面影响。例如,城市化通过共享生活环境、直接接触或人类饮食,导致了郊狼(Canis latrans)肠道微生物组的失调,进一步影响了它们的健康。人类活动引起的栖息地退化和碎片化已导致野生动物肠道微生物α多样性下降,例如Tome's多刺鼠(Proechimys semispinosus)、黑吼猴(Alouatta pigra)、灰褐鼠狐猴(Microcebus griseorufus)和乌德宗瓦山红疣猴(Procolobus gordonorum)。因此,除了传统上关注三个生物多样性层次的野生动物保护观点之外,我们提出了第四个层次:野生动物的共生微生物多样性(比如肠道微生物组)。所提出的“生物多样性第四层级”——共生微生物多样性——不仅仅是一个附加组成部分,更是对宿主适应能力的功能性增强。重要的是,微生物多样性介导了基本的生理过程和在尺度上与遗传、物种或生态系统多样性不同的功能延伸:(1) 营养门户:对于缺乏内源性纤维素酶的叶食性灵长类,肠道微生物组对于将顽固的植物多糖转化为可代谢能量可能是不可或缺的 ,这直接影响了它们在贫营养石灰岩森林中的生存;(2) 环境感应:肠道微生物可能有助于响应栖息地改变(例如,人类食物引起的饮食转变) ,可作为生态系统健康退化的早期预警生物标志物;(3) 进化遗产:宿主-微生物共分化模式表明,宿主特异性的微生物群落可能代表了在宿主谱系进化中保守的一个“扩展基因组” 。因此,保护灵长类共生微生物不仅仅是传统保护工作的附属品,而是在快速变化的环境中保持适应恢复力的机制性必然要求。
目前,分布于新热带地区、非洲大陆、马达加斯加、南亚和东南亚的现存灵长类物种中约有65%面临灭绝威胁,75%的种群数量正在下降,这主要源于全球市场需求增加和工业农业、畜牧业生产及自然资源开采导致的土地转换所引起的森林砍伐和栖息地丧失。因此,考虑到先前研究和我们在野生叶猴中的发现,野生灵长类不仅面临宿主遗传多样性丧失的威胁,也面临共生微生物组变化的威胁。在此,我们提出生物多样性的第四个层次:未来野生动物保护中的共生微生物多样性 。
除了作为人为干扰的标志物,考虑观察到的HDR相关类群富集是否会对叶猴的功能或健康产生后果也很重要。毛螺菌科和普雷沃菌科等科是哺乳动物肠道菌群的常见成员,有助于复杂碳水化合物的发酵和短链脂肪酸的生产 。然而,向高糖低纤维含量的饮食转变与这些类群的显著变化以及人类肠道菌群失调有关 。例如,Prevotella丰度的增加与不同的结果相关,这取决于饮食背景,包括高纤维摄入的有益效果,但在某些条件下也会引发促炎反应 。类似地,在较高干扰水平下富集的Treponema物种可能既包括有助于纤维降解的共生谱系,也包括与其他宿主感染相关的机会性物种 。这些组成变化可能不仅反映了“人源化”,也反映了潜在的健康风险,例如本地发酵途径的中断或从人类或家畜传播病原体的风险增加。然而,我们目前的数据并未评估宿主的健康状况或免疫反应。我们承认,功能验证——如代谢组学、病原体筛查和宿主炎症检测——对于确定这些微生物变化是有害的还是适应性的非常必要。未来的研究应将微生物组分析与宿主生理测量相结合,以更好地理解人为干扰下微生物组“人源化”的后果。
结果和讨论
影响野生叶猴肠道微生物物种的因素
我们发现四个组别之间的肠道微生物组成存在一些差异:广西野生黑叶猴(HYHGX)、广西野生白头叶猴(BTYH)、贵州野生黑叶猴(HYHGZ)以及广西南宁动物园的其他圈养叶猴物种(YHZOO)。这些叶猴中的优势微生物物种包括厚壁菌门(Firmicutes)(例如,Ruminococcaceae UCG014_uncultured bacterium, Ruminococcaceae UCG005_uncultured bacterium, Lachnospiraceae NK4A136 group_uncultured bacterium, Christensenellaceae R-7 group, Eubacterium coprostanoligenes group_uncultured bacterium)、拟杆菌门(Bacteroidetes)(Bacteroidales S24-7 group_uncultured bacterium, Prevotella7_unclassified, Rikenellaceae RC9 gut group_uncultured bacterium)、变形菌门(Proteobacteria)(例如,Pandoraea norimbergensis)和疣微菌门(Verrucomicrobia)(例如,Akkermansia muciniphila ATCC BAA-835)(图2A和2B)。许多瘤胃球菌科和拟杆菌科的物种在碳水化合物代谢(例如,多糖降解)中起重要作用 [27,48,49]。这些常见的微生物物种可能与其草食性饮食(例如,食叶)有关。此外,一些微生物物种的相对丰度在四组之间存在显著差异(图2B)。例如,在前50个物种中,有16个物种(Cellvibrio_unclassified, Oxalicibacterium horti, Erythrobacter attanticus, and Nocardiopsis_unclassified)在HYHGX(广西黑叶猴)中显著富集,4个物种(Treponema 2_unclassified and Desemzia_unclassified)在HYHGZ(贵州黑叶猴)中富集。9个物种(Christensenellaceae R-7 group, Ruminococcaceae UCG014_uncultured bacterium, Ruminococcaceae UCG010_uncultured bacterium, and Campylobacter troglodytes)在BTYH(广西白头叶猴)中显著富集,21个物种(其中大多数属于普雷沃菌科、毛螺菌科和螺旋体科)在YHZOO(圈养叶猴)中富集(图2C)。BTYH成员拥有不同的优势瘤胃球菌科物种。Cellvibrio物种在纤维素降解中起重要作用 [50]。与野生种群相比,圈养非人灵长类中普雷沃菌科和拟杆菌科的一些物种更为富集 [51]。因此,我们推测饮食、宿主系统发育和生活方式等因素可能影响肠道微生物群落。
环境样本(BTYH生活环境中的土壤、水和岩石)具有不同的微生物组成。变形菌门微生物物种(例如,Pandoraea norimbergensis, Phyllobacterium_uncultured bacterium, Brevundimonas intermedia, Achromobacter_unclassified, and Delfia_unclassified)的相对丰度在环境样本中相对高于叶猴粪便样本(图2A和2B)。在这八个组别(包括叶猴和生存环境)中,我们发现了一个核心ASV(图2D),它来自Pandoraea norimbergensis,并且在BTYH栖息地的食物、水和土壤样本中高度丰富(图2E)。该ASV的相对丰度在位于不同山区或城市的HYHGZ和YHZOO中较低(图1A)。使用ASV进行的源追踪分析证实,大约9%的BTYH微生物组可能来自1月(旱季)的水体微生物组(图2F),大约12%的BTYH微生物组可能来自7月(雨季)的食物微生物组(图2G);这种环境微生物组贡献的主要ASV来自Pandoraea norimbergensis(图2H和2I)。Pandoraea norimbergensis已从环境(例如,水和土壤)中分离出来。因此,这些发现进一步证实了生存环境可以影响叶猴肠道微生物组:特殊的地方环境微生物组可能会传播到叶猴肠道微生物组中。有趣的是,环境微生物组的贡献表现出季节差异。在野外观察中,我们发现白头叶猴在旱季的饮水频率高于雨季。在雨季,我们观察到它们的主要水分摄入来源是植物性食物。因此,叶猴的行为可能进一步塑造其肠道微生物组。
最后,使用未加权UniFrac距离的非度量多维尺度分析显示,一个主要由HYHGZ和YHZOO样本组成的聚类,表明它们之间的肠道微生物群落高度相似。然而,HYHGX在肠道微生物群落上表现出显著差异,而BTYH在这些聚类中处于中间位置(图2J)。HYHGZ和HYHGX同属于黑叶猴样本,但生活在不同的山区(图1A)。然而,HYHGZ种群的叶猴居住地靠近村庄,并经常造访人类住区寻找食物(图S2)。基于HYHGX与圈养YHZOO肠道微生物组的高度相似性,以及HYHGZ与HYHGX之间的高度差异性,我们推断人类干扰可能也影响了野生叶猴的肠道微生物组,导致了一些趋同特征。
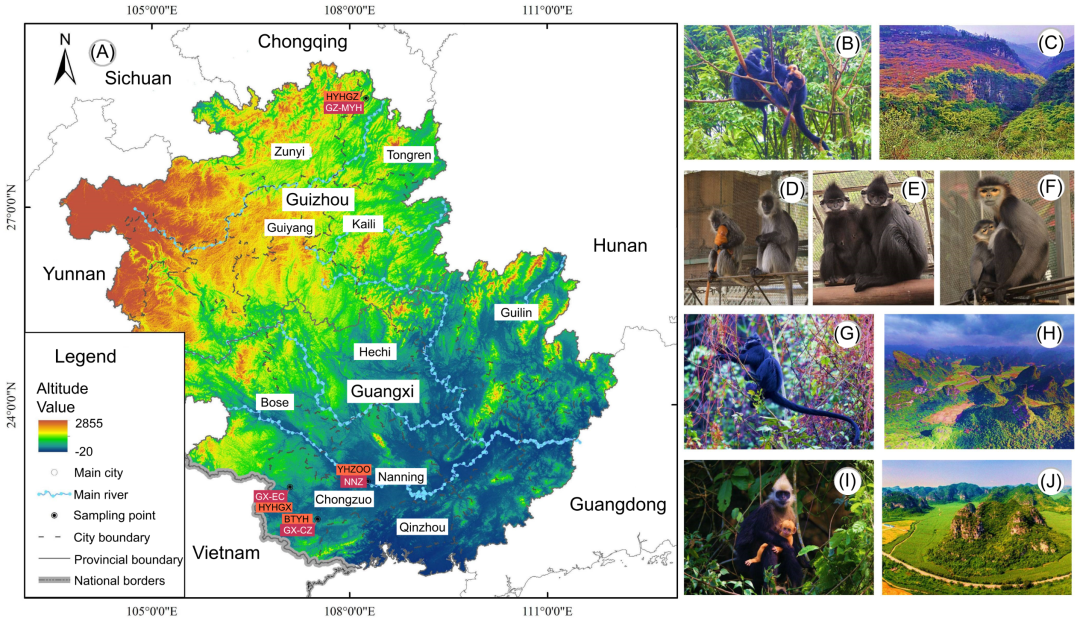
图1. 研究地点和对象:黑叶猴
(A) 主要研究区域分布:研究区域主要集中在中国贵州和广西省,辅以当地地理标志(城市、河流、海拔梯度等,详见图例)以明确采样点的空间分布。(B, C) 贵州麻阳河国家级自然保护区(GZ-MYH组, HYHGZ):(B) 自然栖息地中的黑叶猴;(C) 保护区内喀斯特山地森林景观。(D--F) 南宁动物园(NNZ组, YHZOO):(D) 银叶猴;(E) 黑叶猴;(F) 黑腿白臀叶猴。(G, H) 广西恩城国家级自然保护区(GX-EC组, HYHGX):(G) 黑叶猴;(H) 广西典型喀斯特峰林景观。(I, J) 广西崇左白头叶猴国家级自然保护区(GX-CZ组, BTYH):(I) 白头叶猴;(J) 保护区内森林覆盖的喀斯特山谷景观。
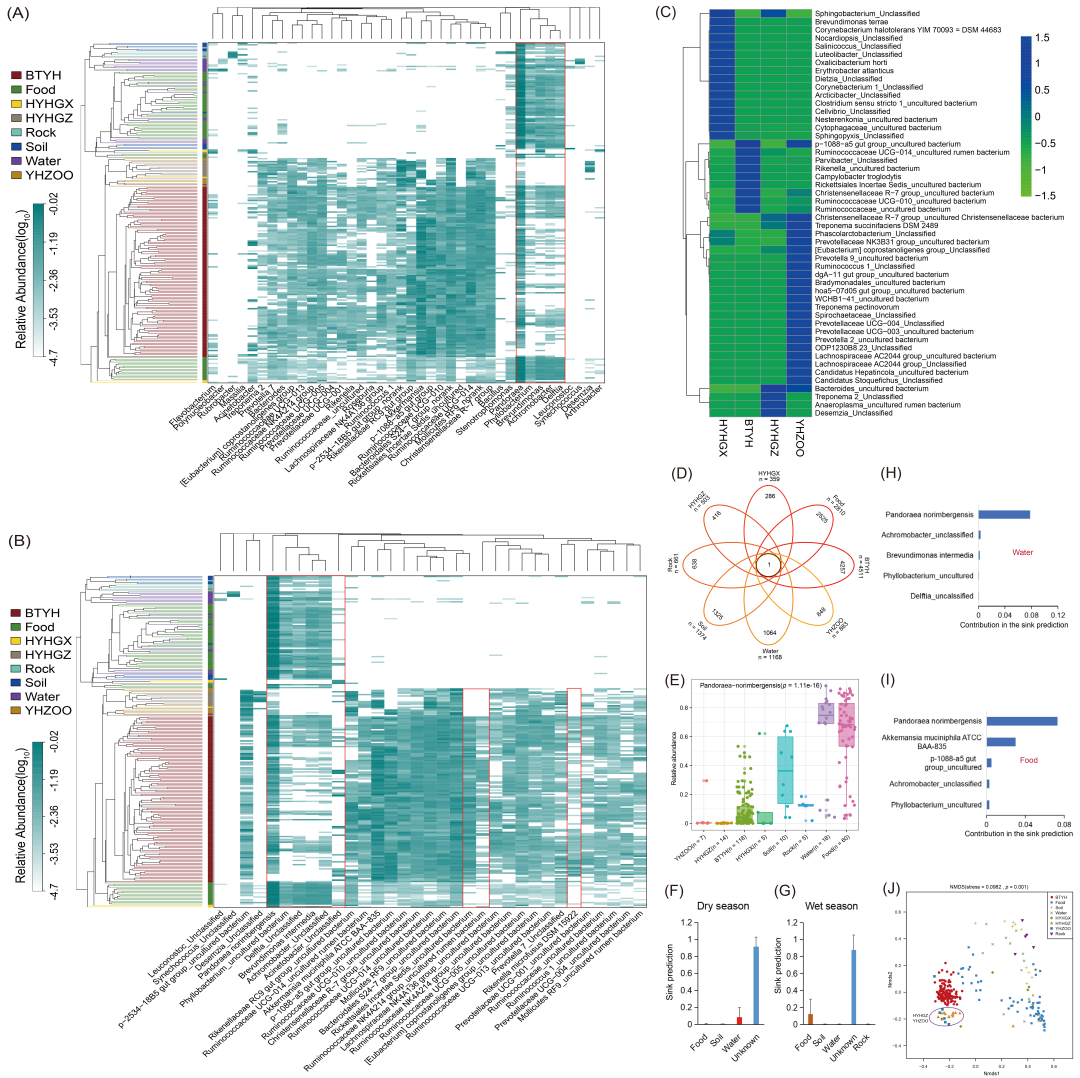
图2. 基于16S全长测序的237个样本肠道微生物组组成和差异的综合概览
(A) 以相对丰度热图显示优势属组成(LG)。(B) 优势种组成,其中大多数物种针对SILVA 132数据库未分类。层次树基于未加权UniFrac距离构建。(C) 经过z-score转换的热图显示各组间前50个物种的相对丰度。这些物种的相对丰度在组间存在显著差异(Kruskal-Wallis检验, p < 0.05)。(D) 维恩图说明八个组之间共享的核心ASV。(E) 八个组中单个核心ASV(Pandoraea norimbergensis)的相对丰度。使用双因素方差分析(Two-sided ANOVA, p = 1.11e-16)检验统计学显著性。(F) 旱季期间环境微生物组对野生白头叶猴(BTYH)肠道微生物组的推定贡献。(G) 雨季期间环境微生物组对BTYH肠道微生物组的推定贡献。(H) 水源微生物组对BTYH肠道微生物组的主要ASV贡献。(I) 食物源微生物组对BTYH肠道微生物组的主要ASV贡献。(J) 基于未加权UniFrac距离的非度量多维尺度分析(NMDS)显示237个样本的肠道微生物组组成。Stress值 = 0.0982。使用双因素置换多元方差分析(Two-sided PERMANOVA, p = 0.001)检验统计学显著性。
人类干扰对肠道微生物的潜在影响:共生微生物组保护
我们发现典型的人类干扰相关(HDR)肠道微生物组的相对丰度与人为干扰程度之间存在正相关关系(图3A−D)。我们使用这些因素(例如,土地利用、村庄和道路)计算了人为干扰,发现从HYHGX到HYHGZ和BTYH,人为干扰增加,BTYH处于中间位置(图3A)。因此,为了展示相关性,我们将HYHGX、BTYH和HYHGZ的人为干扰值分别定义为0.25、0.5和0.75。生活在动物园(YHZOO)的圈养叶猴所受的人为干扰被视为最强的干扰,值为1。四种典型的HDR肠道微生物科(毛螺菌科、Bacteroidales S24-7 group、普雷沃菌科和螺旋体科)的相对丰度随着人为干扰的增加而增加(图3B),这同样发生在典型的HDR属中,例如Lachnospiraceae_uncultured, Bacteroidales S24-7 group_no rank, Prevotellaceae UGC-004, and Treponema 2(图3C)。门、科和物种水平上前30位的相对丰度进一步显示了随着人为干扰增加而发生的深刻变化(图3D)。毛螺菌科的物种在人类肠道微生物组中比例很高,能够形成孢子,便于在宿主间传播 。属于螺旋体科的厌氧Treponema物种是典型的人类肠道病原体。它们的相对丰度在农村人群中尤其高 。Treponema pectinovorum是从牙周炎患者的龈上及龈下样本中分离出来的。Treponema succinifaciens是一种非致病性物种,最初从猪中分离 。在YHZOO样本中发现了属于Treponema pectinovorum的ASV,而在HYHGZ和YHZOO样本中存在属于Treponema succinifaciens的ASV。HYHGZ的成员生活靠近并经常进入村庄。在这些村庄中,某些家畜,如猪、羊和牛,是散养的。我们推测叶猴种群中毛螺菌科和Treponema丰度的增加可能是由于在人类干扰增加期间(例如,在同一生存环境(村庄或动物园)中与人类或家畜的间接和直接接触)传播所致,并随后可能在叶猴社会群体内进一步传播。Prevotella和Bacteroides是现代人类肠道微生物组中的优势属,参与碳水化合物和蛋白质代谢 。十个非人灵长类在圈养(例如,动物园)条件下会被Prevotella和Bacteroides定植,这可能是由于膳食纤维和植物含量减少所致。在野外观察期间,我们记录到HYHGZ的成员经常进入村庄并食用人类食物(包括丢弃的食物)。HYHGZ种群的食物中也含有较高比例的作物,如玉米(图S2)和红薯。除了树叶,圈养叶猴(YHZOO)还进食高比例的水果(例如,香蕉、苹果)、玉米和高粱馒头以及蔬菜,这些每天占其总饮食的20%以上。
因此,我们推测叶猴种群中普雷沃菌科物种和Bacteroidales S24-7 group物种的增加可能是由饮食变化和糖摄入量增加引起的,例如动物园中的玉米高粱馒头、水果,以及HYHGZ中的人类食物、玉米和红薯。然而,这一假设需要通过物种水平的肠道功能分析来验证。近来,人类开发与破坏(例如,森林砍伐、侵占、城市化和食物投喂)已被证明对野生动物的肠道微生物组产生负面影响,例如郊狼、刺鼠、黑吼猴 、灰褐鼠狐猴 、红疣猴和滇金丝猴。在全球层面,约65%的现存灵长类物种面临灭绝威胁,75%的种群数量下降,这主要源于人类干扰,如森林砍伐、土地利用、自然资源开采和城市化。因此,鉴于先前的研究和我们在野生叶猴中的发现,野生灵长类不仅面临宿主遗传多样性丧失的威胁,也面临共生微生物组变化的威胁。在此,我们提出生物多样性的第四个层次:未来野生动物保护中的共生微生物多样性 。
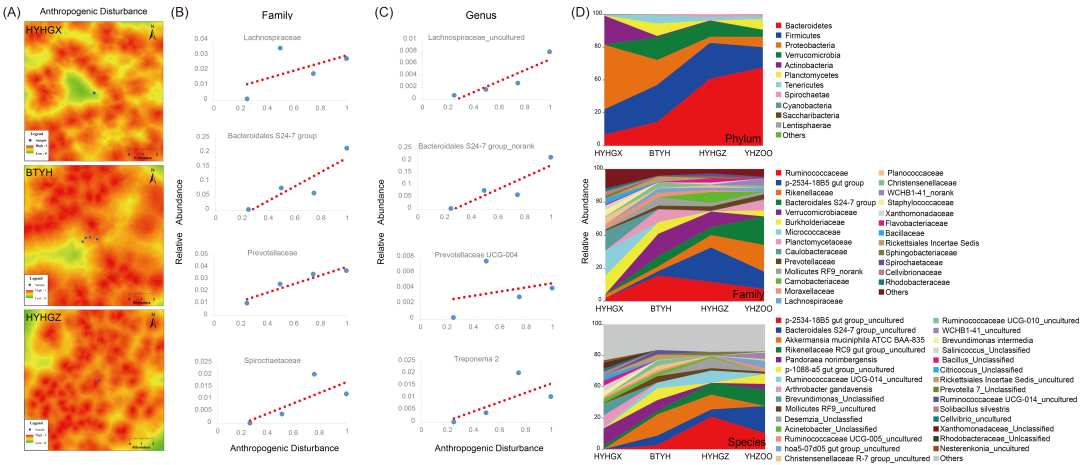
图3. 16S全长测序分析人类干扰与肠道微生物组
(A) 基于关键人为干扰因素(耕地、村庄和交通基础设施如道路)的人类干扰评估。蓝圈表示个体采样点。该评估通过颜色梯度方案可视化,其中红色表示高干扰强度(强度 = 1),绿色表示低干扰强度(强度 = 0)。可视化涵盖三个代表性研究区域:HYHGX(广西恩城)、BTYH(广西崇左)和HYHGZ(贵州麻阳河)。(B−C) 人类干扰相关肠道微生物(科 (B) 和属 (C))在人类干扰下的变化。选择HDR微生物组的基本规则基于典型的人类肠道微生物组。(D) 人类干扰下叶猴肠道微生物组的变化。
1,199个微生物基因组的功能反映了对食叶(高比例多糖)和生存环境的推定适应
本研究中的16S全长测序显示了食叶Presbytis猴的肠道微生物物种组成,尽管大多数未被分类或属于未培养细菌,并揭示了与植物饮食消化相关的推定微生物物种。深度宏基因组测序帮助我们基于比较基因组学分析探索肠道微生物组功能。在此,我们获得了超过2,647个高质量的宏基因组组装基因组(MAGs),覆盖率 > 80%,污染率 < 10%,在去除冗余后最终保留了1,199个非冗余高质量MAGs,平均核苷酸一致性(ANI) ≥ 96%。这1,199个MAGs中的大多数属于厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)和放线菌门(Actinobacteriota),并且是未分类或未培养的细菌(89.5%)(图4A)。大多数变形菌门和放线菌门MAGs的相对丰度在HYHGX中高于其他组(图4B)。大多数变形菌门和放线菌门MAGs在一个模块中显示出正共现关系;然而,它们与厚壁菌门MAGs表现出相互竞争(图S3)。大多数厚壁菌门MAGs显示出正共现关系(图S3)。这1,199个MAGs的分类与16S全长结果一致,并证实了HDR肠道微生物组与人类干扰之间的趋势。例如,当应用基于比对的方法(每个宏基因组中比对上这1,199个MAGs的读数)时,大多数拟杆菌门MAGs的相对丰度在HYHGZ和YHZOO宏基因组中较高(图4A)。毛螺菌科、拟杆菌科(包括NR数据库中的Prevotella)和Treponema的相对丰度随着人为干扰的增加而增加(图4B−4D)。11个MAGs属于Treponema,其中两个属于Treponema succinifaciens,这在本研究的16S全长测序中已鉴定。此外,使用Bray-Curtis距离对这47个宏基因组进行的PCA聚类分析显示,无论是在物种群落(图S4A)还是功能群落(图S4B)上都具有高度相似性,表明了人为干扰的潜在影响。因此,在本研究中,基于深度宏基因组数据(每个宏基因组超过50 G)的分箱(binning)是研究肠道微生物物种组成的一种好方法,结合了高通量16S全长测序。此外,大量MAGs的最大优势是,我们可以在特殊宿主条件(例如,食叶和生活在石灰岩森林中)下探索物种水平的推定肠道微生物组功能。
四个叶猴组(HYHGX, BTYH, HYHGZ, YHZOO)之间优势门和物种的显著差异模式表明,宿主饮食习惯可能在塑造肠道微生物群落中起关键作用(图4A−D)。为研究这一假设,我们使用直接取食观察(针对野生叶猴)和食物摄入记录(针对圈养叶猴)分析了每组的饮食组成(图4E−H)。这些饮食差异可能是观察到的微生物群落分化的基础(图4D),强调了在灵长类肠道微生物组解释中整合饮食信息的必要性。基于这些结果,我们接下来通过分析碳水化合物活性酶(CAZymes)和代谢途径的富集来评估肠道微生物组的功能潜力(图5A)。京都基因与基因组百科全书(KEGG)途径中参与纤维素消化的酶主要包括内切葡聚糖酶(EC 3.2.1.4)、β-葡萄糖苷酶(EC 3.2.1.21)和纤维素1,4-β-纤维二糖苷酶(EC 3.2.1.91)。我们发现了177个同时具有推定EC 3.2.1.4和EC 3.2.1.21的MAGs,以及17个具有EC 3.2.1.91的MAGs(图5A)。这些MAGs主要属于拟杆菌科(Bacteroidaceae)、Muribaculaceae、毛螺菌科(Lachnospiraceae)、瘤胃球菌科(Ruminococcaceae)、Christensenellales_CAG-74和Cellvibrionaceae(表S2)。这177个MAGs中有两个属于Treponema:Treponema_unclassified 和 Treponema succinifaciens。Treponema succinifaciens可以发酵富含纤维的饮食成分。野生白头叶猴饮食中粗纤维的平均比例约为20%(图4E和5B)。因此,我们推断这177个MAGs可能参与纤维素消化。有趣的是,有六个MAGs同时包含这三种酶。这六个MAGs中有五个来自HYHGX样本,其中三个属于变形菌门的Cellvibrio,各有一个属于放线菌门的Nocardiopsis和Cellulomonas。这六个MAGs中有一个来源于BTYH样本,属于变形菌门的Cellvibrionaceae。16S全长分析也显示Cellvibrio_unclassified 和 Nocardiopsis_unclassified 在HYHGX中显著富集。Cellvibrionaceae(包括Cellvibrio属)可以降解多种多糖,包括纤维素。Nocardiopsis和Cellulomonas也能降解纤维素。因此,我们推测这六个MAGs可能在纤维素消化中扮演某种“超级”角色。此外,碳水化合物活性酶数据库(CAZy)中的糖苷水解酶家族5(GH5)包含约21种参与多糖降解的酶,如EC3.2.1.4, EC 3.2.1.21, EC 3.2.1.91, EC 3.2.1.8, 和 EC 3.2.1.45。我们发现GH5存在于这些优势门中(图4A),并且主要富集于属于瘤胃球菌科(包括Ruminiclostridium, Ruminococcus, 和 CAG-115等属)的MAGS中,它们是本研究中叶猴肠道微生物组的优势科。瘤胃球菌科物种在动物和人类肠道微生物组的多糖降解中起作用[78−80]。我们还获得了属于Fibrobacter intestinalis的MAGs(YYF6-1, 在YHZOO中的Bin225),这是一种哺乳动物大肠中的典型纤维素分解菌[80,81]。16S全长测序也在YHZOO组的YYF7样本中发现了两个属于Fibrobacter_uncultured bacterium的ASV。然而,我们未在YYF7样本中获得属于厚壁菌门的MAGs,可能由于这些细菌丰度较低以及分箱过程中的组装偏差。总体而言,我们利用16S全长方法和1,199个MAGs的比较基因组学分析证据,揭示了食叶叶猴中参与潜在膳食来源多糖(例如,纤维素)消化的肠道微生物物种比例很高。这些叶猴灵长类携带了一些推测共有的纤维素分解菌和一些宿主特异性的纤维素分解菌,这可能是由于宿主偏好和生存环境所致。
为了进一步可视化这三种关键酶(EC 3.2.1.4, EC 3.2.1.21, EC 3.2.1.91)在六个MAGs中的分布,我们生成了一个Sankey图(图5C),该图显示Cellvibrionaceae和Ruminococcaceae的MAGs对这三种酶的共同表达贡献巨大。具体来说,Cellvibrionaceae MAGs(例如,Bin225)主导了EC 3.2.1.4和EC 3.2.1.21的共表达,而Ruminococcaceae MAGs(例如,YYF6-1)则富集了EC 3.2.1.91。在此基础上,我们通过系统发育和功能分析比较了Ruminococcaceae MAGs(一个关键的纤维素分解科)的基因组内容(图5D)。结果显示,来自野生叶猴(HYHGX, BTYH)的Ruminococcaceae MAGs比来自圈养YHZOO叶猴的MAGs拥有更完整的纤维素降解基因集(例如,celA, celB),这与它们较高的粗纤维摄入量相一致。
这些纤维素降解微生物可能通过将不可消化的植物多糖转化为可吸收的代谢物来促进宿主适应。具体来说,瘤胃球菌科和毛螺菌科的纤维素分解成员,以及选定的Clostridium物种(例如,C. cellulovorans),编码糖苷水解酶GH5和GH9。这些酶协同将膳食纤维降解为可发酵的寡糖,随后被肠道微生物群代谢为短链脂肪酸(SCFAs),包括乙酸盐、丙酸盐和丁酸盐。SCFAs是主要的能源,其中丁酸盐支持结肠上皮健康,乙酸盐/丙酸盐参与全身代谢调节。这种微生物代谢能力对于叶食性灵长类在低蛋白、高纤维环境中生存至关重要,通过增强能量获取、维持肠道屏障完整性和调节免疫反应来实现。因此,存在一个多样化和功能特化的纤维降解细菌群落,是食叶叶猴饮食适应的关键微生物机制。
哺乳动物基因组不编码许多参与植物多糖降解的酶。哺乳动物肠道微生物对膳食来源多糖和宿主来源聚糖的发酵导致短链脂肪酸的形成,并对健康产生重要影响。例如,尽管大熊猫的基因组没有编码与纤维素和半纤维素消化相关酶的基因,但食竹大熊猫的肠道微生物帮助宿主消化膳食竹子中的多糖。到达哺乳动物肠道的膳食多糖主要成分主要包括植物细胞壁多糖,以及各种寡糖和储存多糖,如菊粉和部分淀粉。在此,我们进一步研究了食叶叶猴肠道微生物物种在消化膳食来源多糖(例如,淀粉、菊粉、果胶、纤维素和阿拉伯木聚糖)和宿主来源聚糖(例如,黏蛋白)中的功能。
使用CAZy数据库和参与这五种类型膳食来源多糖降解的糖苷水解酶(GH)家族(图6A),我们鉴定出167个MAGs,每种降解类型至少有一个GH家族(表S3)。这些MAGs可能在一定程度上参与这五种类型的膳食来源多糖降解。属于每组的这些MAGs的数量随着人类干扰的增加而增加:6个HYYGX深度宏基因组中有10个MAGs,26个BTYH深度宏基因组中有12个MAGs,4个HYHGZ深度宏基因组中有31个MAGs,11个YHZOO深度宏基因组中有167个MAGs(图6B)。这167个MAGs主要属于厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)(图6C)。在科的水平上,它们大多属于Christensenellaceae_CAG-74, Oscillospirale_CAG-272, 毛螺菌科(Lachnospiraceae), 瘤胃球菌科(Ruminococcaceae), 拟杆菌科(Bacteroidaceae), Muribaculaceae, 和 Verrucomicrobiota_UBA1067(图6D)。例如,Christensenellaes_CAG-74是这四组中最常见的科(图4C和图S5),并且许多Christensenellaes_CAG-74 MAGs在CAZy家族数量上富集,并具有中等数量的GH5和EC 3.2.1.21(图S5),表明其在叶猴膳食来源多糖降解中的潜在作用,但纤维素消化能力有限。属于这些HDR毛螺菌科(图6E)和拟杆菌科(包括Prevotella)(图S6和表S3)的MAGs中,CAZy GH家族的数量以及参与KEGG淀粉和蔗糖代谢的酶都很丰富。我们证实了先前的发现,即毛螺菌科在碳水化合物代谢中起重要作用。圈养条件下Prevotella和Bacteroides的增加可能是由于膳食纤维减少所致。因此,我们的研究支持了基于16S全长结果的推测,即叶猴种群中HDR毛螺菌科和拟杆菌科物种的增加可能是由于在人类干扰下HYHGZ和YHZOO的糖摄入增加和植物纤维减少所致(图4E)。
我们鉴定出三个来自疣微菌门(Verrucomicrobiota)的MAGs可能参与宿主来源聚糖,特别是黏蛋白的降解。这些包括Akkermansia MAGs(ZWY2.Bin7: Akkermansia_unclassified; HYF30-1.Bin105: Akkermansia muciniphila)和ECHYM5.Bin45(UBA1315_unclassified)(图6F)。其中,ZWY2.Bin7(Akkermansia unclassified)和HYF30-1.Bin105(Akkermansia muciniphila)在所有叶猴组中都被检测到,但在YHZOO(圈养)组中丰度更高(图6F)。A. muciniphila是一种特征明确的黏蛋白降解细菌,定植于大肠黏液层,在黏膜稳态和宿主聚糖代谢中起重要作用。黏蛋白是一类由大多数动物的上皮组织产生的高分子量、高度糖基化的蛋白质家族。在本研究中,16S rRNA全长数据和宏基因组MAG分析一致显示Akkermansia muciniphila在YHZOO组中富集(图1D-F和图6F)。这种趋势可能反映了圈养饮食条件下微生物底物可用性的转变。与野生叶猴相比,圈养个体消耗的植物源性纤维显著减少,而加工碳水化合物(如玉米和高粱馒头)以及水果的摄入量增加(图4H)。膳食多糖的减少可能导致肠道微生物利用宿主来源的聚糖——特别是黏蛋白——作为替代碳源,从而促进了A. muciniphila的富集。此外,这三个Akkermansia MAGs含有很少的糖苷水解酶(GH5),且降解膳食来源多糖的能力有限,表明它们专门从事黏蛋白代谢而非植物多糖。在对其他非人灵长类(如金丝猴Rhinopithecus roxellana)的研究中也观察到,金丝猴主要依赖富含纤维的植物获取足够的膳食纤维和复杂的植物多糖。然而,在圈养环境中,人为饮食干扰,如水果和加工精料摄入增加,可能导致必需营养素摄入减少。这种饮食转变不仅改变了肠道微生物组组成,也破坏了前肠发酵的微生物平衡。此外,随着恒定营养摄入的减少,金丝猴中Akkermansia的丰度可能会显著增加。这一发现与我们研究中圈养个体Akkermansia高丰度的观察结果一致。这种功能分化可能反映了在圈养低纤维、高糖饮食下的一种生态位适应策略。此外,哺乳动物肠道细菌发酵膳食来源多糖的终产物包括短链脂肪酸、其他有机酸(例如,甲酸盐)、醇类和气体[例如,氢气(H2)和二氧化碳(CO2)]。古菌产甲烷作用(例如,Methanobrevibacter smithii)可以通过去除H2和其他反应终产物来提高多糖发酵的效率。在此,我们从不同组样本中获得了六个古菌产甲烷MAGs:ZWY9.Bin28(Methanobrevibacter_unclassified),HYM12-1.Bin159(Methanobrevibacter smithii),ECHYM6.Bin135(Methanomethylophilus unclassified),YYF7-1.Bin179(MX-02 sp006954405),ECHYM6.Bin109(UBA71_unclassified),和SJC6.Bin162(UBA71_unclassified)(图6G)。因此,在本研究中,我们从上到下鉴定了食叶叶猴中参与多糖降解的推定物种。
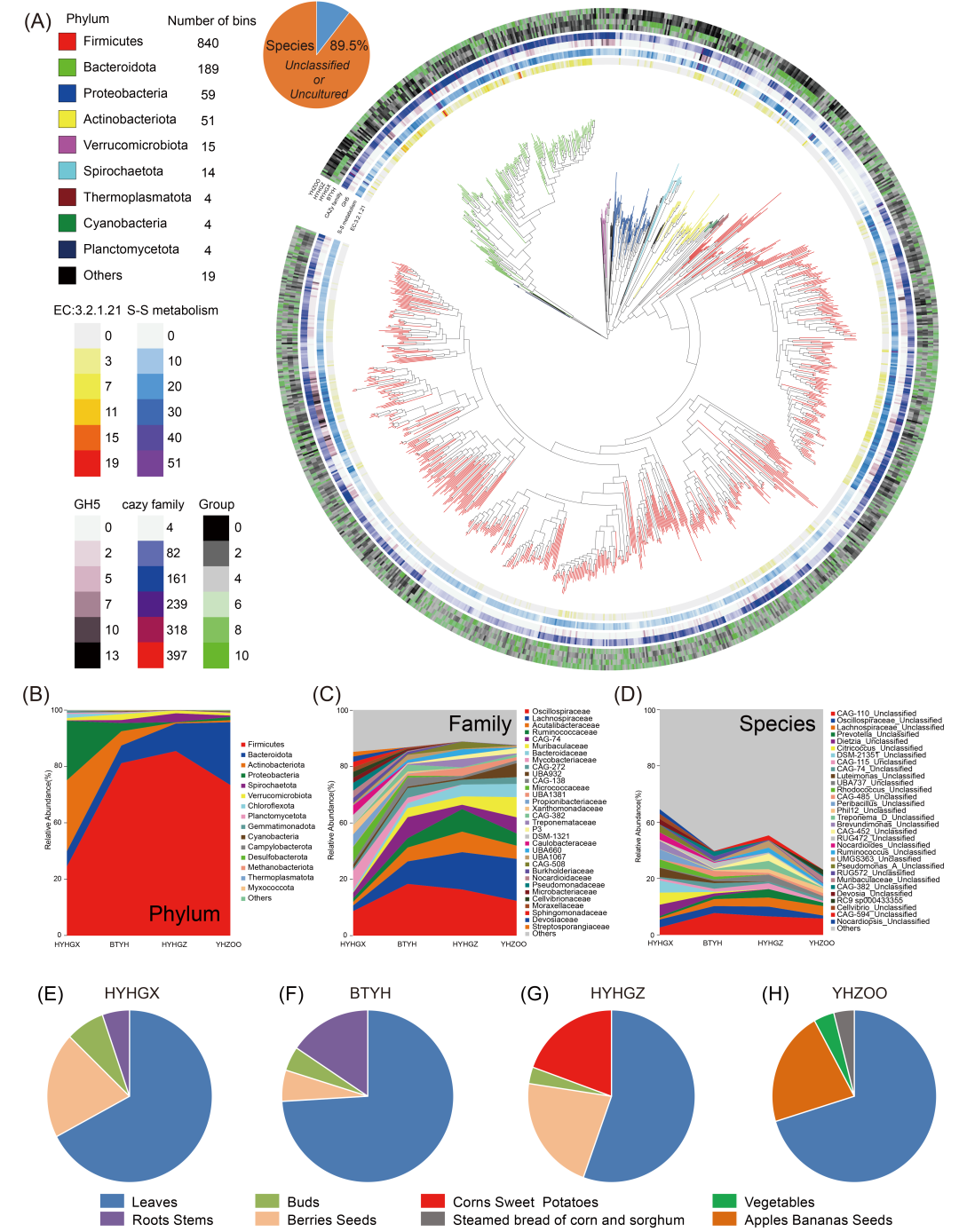
图4. 47个深度宏基因组的1,199个非冗余MAGs的分布
(A) 这1,199个MAGs的系统发育分析。圆圈中心的面板显示使用MAGs创建的最大似然树。外圈热图显示每个bin(MAG)在每个组(HYHGX, BTYH, HYHGZ, YHZOO)或特定通路中的相对丰度。组中的相对丰度:lgTPM(每百万转录本)。EC 3.2.1.21(β-葡萄糖苷酶)、S-S代谢(淀粉和蔗糖代谢)、GH5(糖苷水解酶)、CAZy(碳水化合物活性酶)家族的丰度:基因数量。(B−D) 基于每个宏基因组读段映射到这1,199个MAGs的叶猴各组中优势门、科和物种的相对丰度。(E) 基于取食行为观察的广西野生黑叶猴(HYHGX)饮食组成。(F) 基于取食行为观察的广西野生白头叶猴(BTYH)饮食组成。(G) 基于先前研究重新分析的贵州野生黑叶猴(HYHGZ)饮食组成。(H) 基于食物摄入记录的广西南宁动物园圈养叶猴饮食组成。
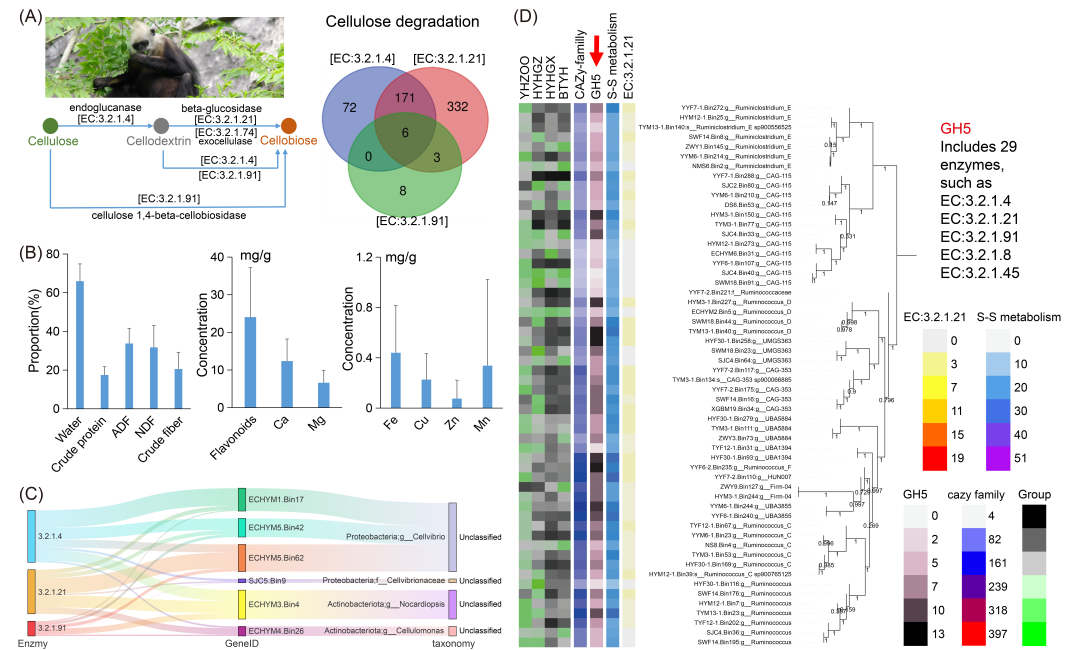
图5. 1,199个叶猴MAGs的比较分析揭示了其在纤维素降解中的推定作用
(A) 维恩图显示了参与纤维素降解的酶的共享信息。纤维素降解途径根据KEGG途径绘制。(B) 广西野生白头叶猴(BTYH)主要膳食植物的营养成分。NDF, 中性洗涤纤维。ADF, 酸性洗涤纤维。(C) Sankey图可视化了对这六个拥有这三种酶的MAGs中参与纤维素降解的这三种关键酶的相对比例。(D) 属于瘤胃球菌科的MAGs的比较分析。
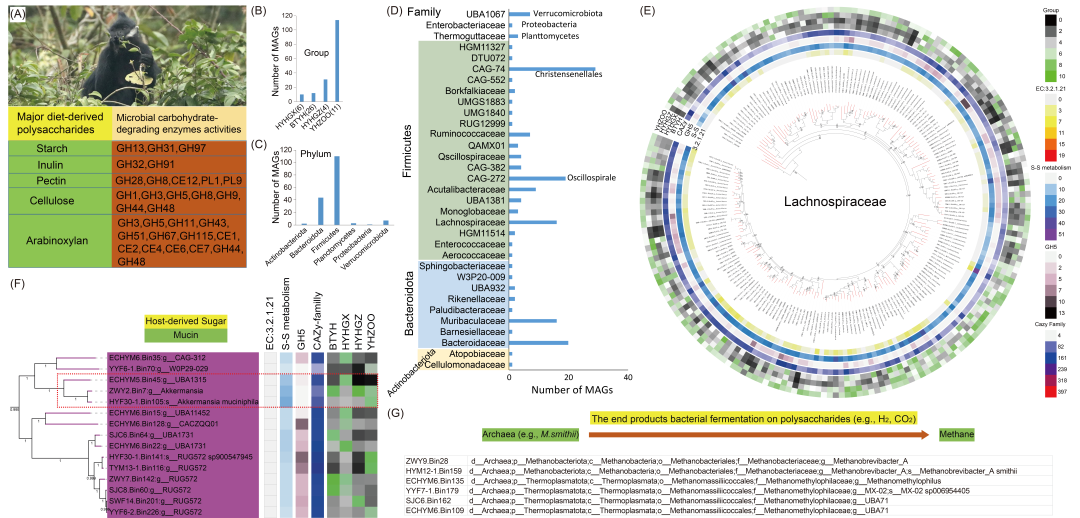
图6. 1,199个叶猴MAGs的比较分析揭示了其在膳食来源多糖和宿主来源聚糖降解中的推定作用
(A) 参与膳食来源多糖降解的CAZy家族。到达哺乳动物肠道的膳食多糖主要成分主要包括植物细胞壁多糖,以及各种寡糖和储存多糖,如菊粉和部分淀粉。(B) 167个MAGs每种膳食来源多糖降解类型至少有一个GH家族。这些167个MAGs的数量属于每组。x轴上的数字代表每组的样本量。这些167个MAGs的数量属于门 (C) 和科 (D)。(E) 属于毛螺菌科的MAGs的比较分析。(F) 参与宿主来源聚糖降解的Akkermansia MAGs的比较分析。(G) 参与细菌发酵终产物降解的古菌MAGs的比较分析。
“石灰岩”猴及其在人为干扰下的草酸盐降解细菌
除了多糖,许多绿叶植物含有高比例的草酸盐和低比例的钙(Ca2+)。植物中Ca2+的浓度范围从1到50 mg/g 。野生白头叶猴膳食叶片中Ca2+的平均浓度约为12 mg/g(图5B)。本研究中的两种野生叶猴栖息于石灰岩森林,其特点是高度多孔和可溶性岩石以及含有高浓度Ca2+的喀斯特地貌。人类高草酸盐水平可导致少数草酸钙结石疾病,如高草酸尿症和肾结石病。哺乳动物酶不编码参与草酸盐降解的酶。哺乳动物清除草酸盐有三种方式:(1) 草酸盐可被吸收到尿路并通过尿液排出;(2) 在肠道中,草酸盐与钙结合形成的不溶性草酸钙可通过粪便排出;(3) 少数肠道微生物可以降解草酸盐,例如Oxalobacter formigenes , Enterococcus faecalis , Providencia rettgeri , Eubacterium lentum, Escherichia coli , 以及Lactobacillus和Bifidobacterium物种。肠道草酸盐降解细菌的缺失或减少会增加高草酸尿症和尿路结石病的风险。因此,研究这些食叶叶猴如何处理叶片中的草酸盐和生存环境中的高浓度钙是值得的。两种细菌酶(Frc: 甲酰辅酶A转移酶 和 Ox: 草酰辅酶A脱羧酶)参与草酸盐降解(图7A)。对这1,199个MAGs的比较分析显示,39个MAGs具有推定的Ox,这些MAGs显示出多样的物种,这可能是由于宿主偏好和生存环境所致(图7B)。例如,39个MAGs中有3个在HYHGX样本中被鉴定,属于两个门:放线菌门(Actinobacteria)(ECHYM4.Bin55: Rhodococcus_unclassified)和变形菌门(Proteobacteria)(ECHYM5.Bin83: Oxalicibacterium_unclassified; ECHYM6.Bin121: Escherichia coli)。有趣的是,我们仅在一个HYHGX样本的两个MAGs中鉴定出推定的草酸盐脱羧酶(OxdD[EC:4.1.1.2])(ECHYM6.Bin150: 厚壁菌门Solibacillus_unclassified; ECHYM6.Bin27: 厚壁菌门Peribacillus_unclassified),它催化草酸盐转化为CO~2~和甲酸盐。这些酶的相对丰度在HYHGX宏基因组中最高(图7C)。Ox的相对丰度在YHZOO宏基因组中最高(图7D)。
结合来自不同哺乳动物类群的87个已发表宏基因组(16个大熊猫(GP),6个小熊猫(RP),19个肉食性食肉动物(CA),10个杂食性食肉动物(OC),12个草食动物(HE),和24个滇金丝猴(16个来自有食物投喂的DJSHF,8个来自无食物投喂的DJSHW)),Rhodococcus和Oxalicibacterium的相对丰度在HYHGX宏基因组中在这11个类群中最高(图7E和7F)。在BTYH样本中鉴定出的39个MAGs中有6个属于三个门:3个MAGs属于Oxalobacter_unclassified(变形菌门),2个MAGs属于Muribaculaceae(拟杆菌门),1个MAG属于ZJ304_unclassified(放线菌门)。Oxalobacter的相对丰度在BTYH宏基因组中在这11个类群中最高(图7G)。在HYHGZ样本中鉴定出的39个MAGs中有7个属于两个门:5个MAGs属于Muribaculaceae(拟杆菌门),1个MAG属于P3(拟杆菌门),1个MAG属于Adlercreutzia(放线菌门)。在YHZOO样本中鉴定出的39个MAGs总共有21个属于三个门:18个MAGs属于Muribaculaceae,1个MAG属于Phocaeicola_unclassifed(拟杆菌门),1个MAG属于Bifidobacteriaceae_unclassified(放线菌门),1个MAG属于Oxalobacter_unclassified(变形菌门)。这三个细菌属Muribaculum(Muribaculaceae科)、Paramuribaculum(Muribaculaceae科)和Phocaeicola的相对丰度随着四个叶猴组人为干扰的增加而增加:在YHZOO宏基因组中最高(图7H−7J)。这种模式在野生滇金丝猴中也存在;DJSHF(有食物投喂)的相对丰度高于DJSHW(无食物投喂)宏基因组(图7H−7J)。Muribaculaceae的物种主要从小鼠肠道中分离出来。圈养或农村环境可能增加了Muribaculaceae物种在叶猴和小鼠之间传播的机会。我们推测,在人为干扰下增加碳水化合物的饮食变化导致了拟杆菌门和Muribaculaceae物种丰度的增加,并进一步受到叶片饮食中草酸盐的刺激。因此,考虑到每个叶猴组中特定的草酸盐降解细菌以及随着人为干扰增加而增加的草酸盐降解Muribaculaceae,我们进一步证实了宿主和生存环境都可以塑造肠道微生物物种的组成和功能。
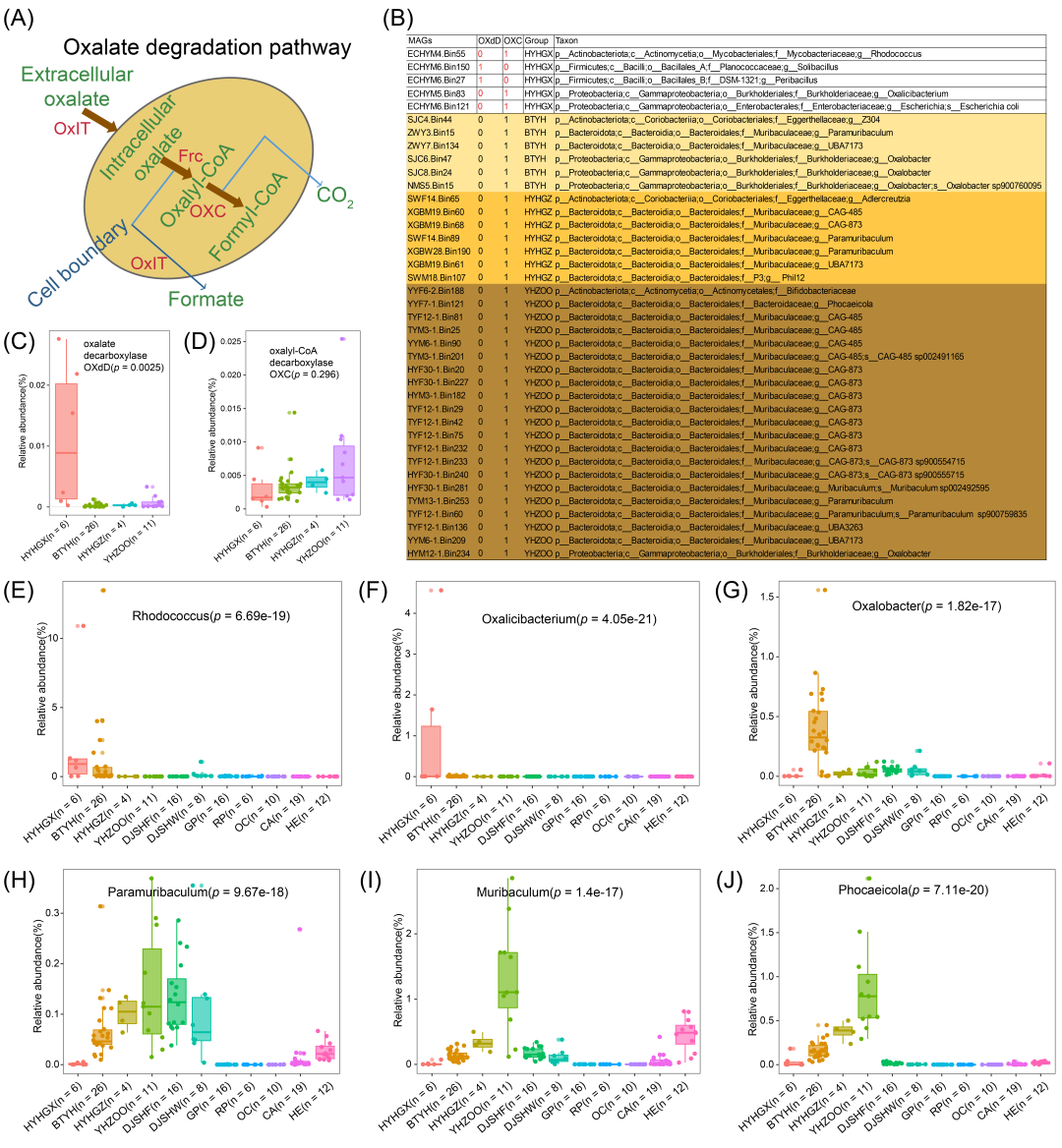
图7. 对1,199个叶猴MAGs的比较分析揭示了其在草酸盐降解中的推定作用
(A) 草酸盐降解途径(根据先前研究重绘)。Frc: 甲酰辅酶A转移酶。Ox: 草酰辅酶A脱羧酶。(B) 39个具有推定Ox的MAGs,这些MAGs显示出多样的物种,这可能是由于宿主偏好和生存环境所致。数字代表编码该酶的基因数量。(C−D) 使用47个宏基因组的叶猴各组中草酸盐脱羧酶(OXdD)和OXC的相对丰度。Kruskal-Wallis检验: p = 4.01e-6 和 p = 0.115。(E−J) 哺乳动物类群间参与推定草酸盐降解的主要细菌科和属的相对丰度。Kruskal-Wallis检验p值: (E) 0.019, (F) 2.48e-5, (G) 5.64e-15, (H) 4.4e-12, (I) 1e-15, (J) 1e-15。我们结合了已发表的87个宏基因组进行分析:16个大熊猫(GP),6个小熊猫(RP),19个肉食性食肉动物(CA),10个杂食性食肉动物(OC),12个草食动物(HE),和24个滇金丝猴(16个来自有食物投喂的DJSHF,8个来自无食物投喂的DJSHW)。
结 论
在此,我们利用来自237个样本的16S全长测序和1,199个微生物基因组,探索了食叶叶猴在细菌物种水平上的肠道微生物组成和功能,并揭示了在碳水化合物和草酸盐代谢降解方面的复杂物种谱。我们得出结论,饮食、宿主系统发育和生存环境可能影响肠道微生物群落和功能。叶猴肠道微生物组在帮助宿主适应叶片饮食和石灰岩生存环境方面发挥着潜在作用。此外,我们揭示了HDR微生物丰度增加与人为干扰增加之间存在潜在的正相关关系。鉴于已知人为干扰对动物肠道微生物组的负面影响,以及全球层面大多数灵长类因人类活动而受威胁的现状,我们提出了生物多样性的第四层级:未来野生动物保护中的共生微生物多样性。未来,野生灵长类的保护将不仅关注宿主遗传多样性,还将关注共生微生物组的变化。
方 法
研究地点
崇左白头叶猴国家级自然保护区(分析中的GX-CZ即BTYH组,22°24′--22°46′N, 107°22′--107°42′E)和恩城黑叶猴国家级自然保护区(分析中的GX-EC即HYHGX组,22°36--22°49N, 106°58′12--107°15′45" E)位于中国广西西南部(图1A)。两个保护区均由石灰岩丘陵组成,海拔在300至700米之间。有两个 distinct 的季节。雨季通常发生在4月至9月,月降雨量> 50毫米。两个保护区的栖息地特征为季节性石灰岩雨林。然而,由于人类活动,两个保护区现有的栖息地已高度碎片化,石灰岩山丘上的植被受到严重干扰。
麻阳河国家级自然保护区(GZ-MYH组HYHGZ,28°37′--54°20′N, 108°3′--109°45′E)位于中国贵州省东北部,属亚热带湿润气候区,季节分明,分为春季(3月至5月)、夏季(6月至8月)、秋季(9月至11月)和冬季(12月至2月)。该保护区地形以喀斯特山脉和陡峭山谷为特征,海拔在420至1,067米之间。植被为中亚热带常绿阔叶林;然而,除核心保护区外,大部分地区因耕作而广泛碎片化。缓冲区内的原始森林在陡坡上保持完好。
为提高读者对我们多维实验策略的理解,我们提供了一个示意图,总结了整体研究设计,包括地点选择、样本收集、实验室分析和整合数据解读(图S7)。该流程图简洁地可视化了每个方法学组成部分在更广泛研究框架内的相互联系。
景观分析:人为干扰评估
本研究选取了野生猕猴种群BTYH、HYHGX和HYHGZ作为研究对象。同时选取三类主要人类活动——耕地、村落及交通设施(如道路)作为影响野生猕猴的人为干扰因子。通过地理信息系统软件对三类影响因子进行量化处理,并采用加权平均法进行综合评估。本研究中各因素定义如下:(1)农田:基于森林资源清查数据,提取农田斑块的表面矢量图层;(2)村落与交通: 基于林业系统开展的森林资源调查卫星影像数据,结合实地考察,通过卫星影像解译获取村落分布点矢量图层及交通道路分布线矢量图。村落数据选取标准为居住人口达50人的聚居点。交通数据选取标准为路基宽度超过3.5米的开放道路。卫星影像获取时间记录于2019年。
使用ArcGIS软件中的欧几里得距离函数,计算到上述三个人为干扰因素的欧几里得距离作为指标,获得这三个影响因素对叶猴影响程度的定量数据,以栅格数据的形式反映。栅格数据的单位,即值(cell),是到影响因素的最近距离。栅格值越大,表示站点到研究范围内某个影响因素的距离越大,该因素对黑叶猴的影响越小。
为减少不同因素间距离值对数据分析的影响,我们对三个因素的栅格数据进行了归一化,其中每个因素栅格层的中位像元值除以该因素层中的最大值。距离值用于获得值范围在0到1之间的三个影响因子栅格层。根据上述方法获得的欧几里得距离归一化的三个影响因子栅格数据,我们以黑叶猴样本的采样位置为中心,2公里为半径。我们计算了该范围内每个影响因子的加权平均值。选择这个2公里缓冲区是为了近似所研究叶猴群的典型家域大小,基于先前的野外观察和这些石灰岩森林的生态研究。通过在这个半径内取平均值,我们旨在更好地反映它们日常活动范围内实际经历的干扰水平,而不是单一点上的干扰。村庄、耕地和交通这三个影响因子的权重分别赋为0.4、0.4和0.2。这些权重值基于灵长类动物保护及喀斯特景观研究中的前期生态影响评估确定。村庄与农田因其持续的大面积占用及长期的人类活动(如土地清理、牲畜放牧、人类活动)而被赋予更高权重;道路虽具破坏性,但在陡峭的喀斯特地形中通常影响范围更局限且呈线性分布,且核心猕猴栖息地往往难以通过道路抵达。最终,针对每个黑叶猴样本点周边,综合评估了村落、耕地和交通三大影响因素对黑叶猴的综合影响评分。评分范围为0至1,分数越高表明三大影响因素对叶猴的综合影响越大。本研究将圈养环境(如动物园条件)视为最强人类干扰(取值1)。动物园环境被赋予1的干扰值,以体现人为干扰的概念性极限——因其存在持续直接的人工管理、高度改造的定量投喂饮食,以及严重受限的人工化生活空间。此类圈养条件已知会持续重塑非人灵长类动物的肠道微生物组,使其趋向人类样态。
粪便样本收集
我们使用非侵入性方法在野外收集了白头叶猴和黑叶猴的粪便样本。叶猴利用石灰岩悬崖上的洞穴和平台睡觉。清晨从这些睡觉平台收集新鲜粪便样本。所有粪便样本至少间隔1米收集,以确保来自不同个体。对跟踪的叶猴在其排便后立即收集粪便。使用无菌收集管收集样本,立即在干冰中冷冻,运输到实验室,并储存在-80℃冰箱中,直至准备测序文库。
总共从广西崇左(GX-CZ)的白头叶猴(BTYH)收集了125份粪便样本,包括2020年9月来自5个邻近叶猴群的50份样本(植物园站点, n = 12; 弄水站点, n = 9; 独山站点, n = 10; 山加村站点, n = 9; 弄蒙山站点, n = 10),2021年1月来自两个群的35份样本(植物园站点, n = 28; 独山站点, n = 7),以及2021年7月来自一个群的40份样本(植物园站点, n = 40)。广西恩城(GX-EC)的黑叶猴种群数量约为100只。GX-EC黑叶猴的睡觉地点位于陡峭悬崖的洞穴中,使得收集粪便困难。因此,我们仅在2021年3月收集了6份黑叶猴(HYHGX)粪便样本。在贵州麻阳河(GZ-MYH),收集了14份黑叶猴粪便样本,包括2021年4月来自两个站点的4份样本(石望洞站点, n = 2, 响沟坝站点, n = 2),和2021年12月来自后王洞站点的10份样本。此外,我们从南宁动物园(NNZ)的11只个体(属于三种疣猴物种)收集了11份粪便样本,包括3份黑叶猴样本、5份银叶猴(Trachypithecus cristatus)样本和3份黑腿白臀叶猴(Pygathrix nigripes)样本。
取食行为数据收集与分析
饮食数据收集自2019年7月至2020年6月期间广西崇左(GX-CZ)的一群白头叶猴和广西恩城(GX-EC)的一群黑叶猴。每天,数据收集从首次遇到叶猴开始,到它们消失或进入睡觉地点结束。我们使用瞬时扫描取样法进行行为数据收集,每15分钟间隔一次。我们将每次时间间隔内的扫描采样限制在前5分钟。每次扫描期间记录每个个体的活动。为避免对特定个体或特定年龄-性别等级的采样偏差,我们在一次扫描中尽可能收集不同个体的行为记录,以包括研究群中的所有个体并避免重复。当动物取食时,记录它们消耗的植物部位(例如,叶、果实、花和种子)。
在研究期间,我们收集了3726条白头叶猴的取食记录和2477条黑叶猴的取食记录。不同植物部位在总饮食中的百分比表示为它们占取食总记录的百分比。
环境样本收集
为分析食物营养成分对白头叶猴肠道微生物组的影响,我们根据植物园和山加村群饮食组成的数据,于2021年1月收集了前10种食物物种的膳食植物样本。每组收集三十份叶片样本(每个物种3份样本)。为进行比较,我们还从五种主要植物物种收集了样本。然而,这些植物仅贡献了总饮食的< 0.5%。新鲜样本重量从20到50克不等,样本被干燥至恒重。干样本放入适当标记的密封塑料袋中进行营养分析。为分析环境因素对白头叶猴肠道微生物组的影响,我们在广西崇左(GX-CZ)收集了植物、土壤、岩石和水的样本。膳食植物样本于2021年2月和7月收集。每个月,我们从20种主食物种收集40份叶片样本(每种植物2份样本)。我们收集的大多数叶片样本来自叶猴消耗的植物。由于原始植物难以接近或无法获得,少数样本从取食植物附近的其他植物收集。每个样本收集 > 50克植物叶片,并用物种名称标记后密封在密封袋中。我们还从叶猴家域内的主要取食和休息地点收集了土壤、岩石和水样本。2021年2月总共收集了30份样本,包括20份土壤样本和10份水样本。我们还从2021年7月收集了24份样本,包括10份土壤样本、8份水样本和6份岩石样本。所有样本至少间隔50米收集以确保独立性。土壤和岩石样本(每样本> 50克)使用无菌收集管收集,水样本(> 450 mL)存储在瓶子中。所有样本在收集后立即用干冰冷冻,运输到实验室,并储存在-80℃冰箱中,直至准备测序文库。
营养分析
样品干燥前后的重量用于计算样品中的水浓度。样品的水含量以水重百分比表示,使用以下公式:水 (%) = (湿重 - 恒重)/湿重 × 100% (湿重: 鲜重, 恒重: 干重) 。(1) 粗蛋白(CP)含量使用凯氏定氮法测定。粗纤维(CF)、中性洗涤纤维(NDF)和酸性洗涤纤维(ADF)使用自动纤维分析仪采用酸碱消化法测量 [133]。CP、CF、NDF和ADF含量表示为干物质中特定营养成分重量的百分比。此外,类黄酮含量使用比色法测量 。矿物质含量(Ca, Mg, Fe, Cu, Zn, Mn)使用原子吸收光谱法测定。类黄酮和矿物质含量表示为每克干物质的微克数(mg/g)。所有样本在教育部珍稀濒危动物生态与环境保护重点实验室(广西师范大学)进行了三次测试以确保准确性。
16S rRNA基因全长测序
使用MP BioFecal DNA Kit(上海,中国)从每个新鲜粪便样本中提取总微生物DNA。使用MP Bio Soil DNA Kit(上海,中国)从每个环境样本中提取总微生物DNA。我们使用引物27F (5′-AGRGTTYGATYMTGGCTCAG-3′) 和1492R (5′-RGYTACCTTGTTACGACTT-3′) 以及通用细菌条形码来扩增全长16S rRNA基因序列。聚合酶链式反应(PCR)(总体积30 µL)包含15 μL of 2× Gflex PCR buffer, 0.6 μL of Tks Gflex DNA polymerase (1.25 U/μL), 1 µL of 正向引物 (5 pmol/μL), 1 µL of 反向引物 (5 pmol/μL), 50 ng of 模板DNA, and 12.4 µL of PCR级水。PCR设置如下:94 °C 5分钟,随后30个循环的94 °C 30秒,56 °C 50秒,和72 °C 60秒;最后在68 °C延伸5分钟。使用GeneTools Analysis Software (version 4.03.05.0, SynGene) 计算PCR产物浓度,并根据等质量原则确定每个样本所需体积。然后将每个PCR产物混合。我们使用HiPure Gel Pure DNA Mini Kit(广州,中国)回收PCR产物(目标插入片段大小为2 kbp)。按照16S Amplification SMRTbell Library Preparation Kit (Pacific Biosciences, Menlo Park, CA, USA) 的标准程序构建扩增子文库(250 bp 至 < 3 kbp),并使用PacBio Sequel II平台进行测序。
使用QIIME2进行16S rRNA基因全长原始数据分析
我们使用SMRT Link v8.0(默认参数:minPasses ≥ 5, minPredictedAccuracy ≥ 0.9)(PacBio)获得环状共识序列(CCS)。我们使用Lima v1.7.0,基于严格的条形码匹配,获得每个样本的原始CSS。使用基于滑动窗口(20 bp)的Trimmomatic软件对原始CCS序列进行预处理以进行质量控制。质量控制措施如下:(1) 检测并去除模糊碱基,(2) 丢弃平均质量得分低于20的低质量序列。修剪后,使用QIIME2中的DADA2 对清洁的双端读段进一步去噪,之后获得ASV表和代表性序列(约1,000--1,600 bp)。将所有代表性的全长16S rRNA基因序列与SILVA参考数据库(版本132;http://www.arb-silva.de/)进行注释和比对。因此,获得了具有分类学信息的ASV相对丰度水平的表格。最后,我们从270个样本中的237个获得了全长16S rRNA基因(表S1)。通过将ASV表稀疏化到所有237个样本中观察到的最小序列数(5,000),对具有不同测序深度的样本进行标准化。
为鉴定显著富集的微生物类群并评估群落组成的差异,我们采用了适用于微生物组数据的非参数统计方法,考虑了组成的性质和不等的组大小。两组间的 pairwise 比较使用Wilcoxon秩和检验,而多组比较(例如,HYHGX, BTYH, HYHGZ, and YHZOO)使用Kruskal-Wallis检验。所有p值均经过Benjamini-Hochberg错误发现率(FDR)校正以进行多重检验。统计学显著性定义为FDR校正后的p < 0.05。这些分析在R (v4.1.0) 中使用vegan和stats包进行。对于前50个微生物物种,我们使用R生成相对丰度(z-score标准化)的热图 。维恩图 (https://en.wikipedia.org/wiki/Venn_diagram) 用于显示八个组(四个叶猴组和其栖息地中的四个环境组:土壤、水、岩石和膳食植物)之间共享的ASV。为调查环境对BTYH叶猴肠道微生物组的潜在贡献,我们使用了SourceTracker,将BTYH视为汇,环境样本视为源。此外,基于未加权UniFrac距离 ,使用非度量多维尺度(NMDS)和主坐标分析(PCoA)可视化组间的β多样性模式。
典型人类干扰相关(HDR)微生物组的定义
选择HDR微生物组的基本规则基于典型的人类肠道微生物组:根据SILVA分类,三个HDR微生物科(毛螺菌科、Bacteroidales S24-7 group和普雷沃菌科)。在NCBI微生物非冗余(NR)数据库中,拟杆菌科包括Bacteroidales S24-7 group和普雷沃菌科。第四个HDR科是螺旋体科,根据SILVA分类,它包括典型的Treponema物种。根据NCBI微生物非冗余(NR)数据库,Treponema物种属于密螺旋体科(Treponemataceae)。选择此有两个原因:(1) 属于螺旋体科的厌氧Treponema物种是典型的人类肠道病原体。Treponema的相对丰度在农村人群中特别高。(2) Treponema succinifaciens是一种非致病性物种,最初从猪中分离 。HYHGZ的个体生活靠近并经常进入村庄。这些村庄中的一些家畜(例如,猪、羊和牛)是散养的。
深度宏基因组测序与分析
我们从每个叶猴组随机选择47个样本(HYHGX中6个,BTYH中26个,HYHGZ中4个,YHZOO中11个)用于在Illumina HiSeq-PE150平台上进行深度宏基因组测序。每个宏基因组的原始数据约为50-60 G,本研究获得了近3 T的原始读段。深度宏基因组测序的最大优势是可以在微生物基因组水平进行下游分析。此外,为了更深入地研究不同的哺乳动物,我们结合了我们已发表的87个宏基因组进行分析:16个大熊猫(GP) ,6个小熊猫(RP) ,19个肉食性食肉动物(CA) ,10个杂食性食肉动物(OC) ,12个草食动物(HE) ,和24个滇金丝猴(16个来自有食物投喂的DJSHF,8个来自无食物投喂的DJSHW)。使用Cutadapt基于默认参数过滤原始读段。基于黑叶猴基因组(PRJNA488530)和川金丝猴基因组(ASM756505v1)去除宿主污染。使用MEGAHIT组装清洁读段 。使用MetaGeneMark预测contigs的编码区,然后使用CD-HIT进行聚类 。因此,我们获得了unigenes。使用每百万转录本(TPM)值,根据使用bowtie2比对的读段数量来估计unigenes的丰度。使用Diamond 将unigenes与NCBI微生物非冗余(NR)数据库(包括细菌、真菌、古菌和病毒)进行比对。我们仅保留属于细菌的unigenes用于下一步分析。随后,获得了这些细菌unigenes的分类学信息。使用KEGG数据库 对unigenes进行功能注释。使用Bray-Curtis距离对这47个叶猴宏基因组进行的PCA聚类分析显示,在物种和功能群落上均存在聚类模式。
使用MAGs(宏基因组组装基因组)进行比较基因组学分析
去除宿主污染后,每个叶猴宏基因组的清洁读段用于宏基因组组装分析。我们使用Burrows-Wheeler Aligner算法和SAMtools将清洁读段映射到contigs。使用MetaBAT2运行分箱(binning)。然后,我们根据每个宏基因组的映射结果获得每个bin的contigs。此外,使用CheckM评估每个bin的完整性和污染,特别是宏基因组组装基因组(MAG)。我们选择完整性≥70%且污染<10%的bins作为合格的MAGs进行下游分析,遵循广泛接受的质量控制标准。在去除冗余后(平均核苷酸一致性(ANI) ≥96%),我们保留了非冗余高质量bins。最后,使用Salmon 将清洁读段映射到这些高质量bins,并确定每个宏基因组中bins的TPM。使用Diamond 将这些非冗余bins与KEGG通路数据库进行比对。使用HMMScan (http://hmmer.janelia.org/search/hmmscan) 将这些非冗余bins与碳水化合物活性酶(CAZy)数据库进行比对。因此,我们获得了每个bin的KEGG基因和糖苷水解酶(GH)家族组成。因此,我们对编码参与特定代谢途径(例如,多糖)的推定酶的基因进行了比较基因组学分析。使用Prodigal针对GTDB数据库 (http://gtdb.ecogenomic.org/) 获得MAG的分类。使用PhyloPhlAn 为高质量bins构建最大似然树。
MAGs共丰度网络的构建与可视化
为构建微生物共丰度网络,将每个样本的清洁读段映射到1,199个高质量MAGs,以计算它们在样本中的相对丰度。基于这些丰度谱,计算 pairwise Spearman相关系数和相应的p值。保留绝对Spearman相关系数≥ 0.8且FDR校正后p值≤ 0.05的MAG对作为显著共变。总共有853个MAGs符合此标准并被纳入网络。值得注意的是,保留了四个古菌MAGs(3个来自Thermoplasmatota,1个来自Methanobacteriota)。使用Cytoscape (v3.9.1) 可视化网络。在图中,每个节点代表一个MAG,节点颜色反映其门,节点大小表示其在所有样本中的平均丰度,边代表显著相关性(绿色表示正相关,红色表示负相关)。
代码和数据可用性:
序列原始数据已提交至NCBI PRJNA994677 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA994677) 和 PRJNA997075 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA997075)。分析所用的数据和脚本可在 https://github.com/mkchenhua/Zhou2025IMO/ 找到。补充材料(图、表、图文摘要、幻灯片、视频、中文翻译版本和更新材料)可在在线DOI或iMeta Science http://www.imeta.science/imetaomics/ 找到。
引文格式:
Qihai Zhou, Qiuyan Guo, Xinyuan Cui, Tao Meng, Zhuting Pang, Song Wang, Hua Chen, et al. 2025. “Species-level exploration of the gut microbiome in the leaf-eating Presbytis monkeys reflected the effects of anthropogenic activity and specialized dietary niches: conservation on the fourth biodiversity level.”iMetaOmics 2: e70051. https://doi.org/10.1002/imo2.70051.
作者简介

周岐海(第一/讯作者)
● 广西师范大学生命科学学院教授。
● 研究方向为灵长类行为生态和保护生物学,以第一作者和通讯作者在Science、Biological Conservation、Zoological Research、Genome Biology and Evolution、American Journal of Biological Anthropology等期刊发表SCI论文51篇。

郭秋艳(第一作者)
● 广西师范大学生命科学学院研究生(已毕业)。
● 研究方向为灵长类行为生态学,经第一作者在兽类学报发表论文1篇。

崔新远(第一作者)
● 南京中医药大学医学院在读博士研究生。
● 研究方向为濒危野生动物保护的多组学整合研究。以第一作者(含共同)在Evolutionary Applications、Ecology and Evolution和Microorganisms等期刊发表研究论文。

孟涛(第一作者)
● 广西壮族自治区林业勘测设计院,博士,高级工程师。
● 研究方向为濒危野生动物保护生物学及自然保护地规划。主持广西自然科学基金1项、国家林业和草原局科技项目多项,在Zookeys、communications Biology等期刊发表论文7篇。

朱立峰(通讯作者)
● 南京中医药大学医学院教授,博士生导师。
● 研究方向为中医药机制多组学研究:(1)肿瘤发生与演化机制的多组学研究(如基因组学、单细胞-时空转录组、空间蛋白组、微生物组、代谢组学);(2)微生物-肠-脑轴调控神经、代谢系统疾病机制研究及中医药干预策略。先后主持国家级项目6项,相关研究成果在PNAS、Nature Genetics、Current Biology、Science China Life Sciences、mBio、Ecology、Molecular Ecology等期刊发表70余篇。
更多推荐
(▼ 点击跳转)
iMeta | 引用20000+,海普洛斯陈实富发布新版fastp,更快更好地处理FASTQ数据
iMeta | 兰大张东组:使用PhyloSuite进行分子系统发育及系统发育树的统计分析
iMeta | 唐海宝/张兴坦-用于比较基因组学分析的多功能分析套件JCVI
iMeta封面

1卷1期

1卷2期

1卷3期

1卷4期

2卷1期

2卷2期

2卷3期

2卷4期

3卷1期

3卷2期

3卷3期

3卷4期

3卷5期

3卷6期

4卷1期

4卷2期

4卷3期

4卷4期
iMetaOmics封面

1卷1期

1卷2期

2卷1期

2卷2期

2卷3期
iMetaMed封面

1卷1期

1卷2期
期刊简介
“iMeta” 是由威立、宏科学和本领域数千名华人科学家合作出版的开放获取期刊,主编由中科院微生物所刘双江研究员和荷兰格罗宁根大学傅静远教授担任。目的是发表所有领域高影响力的研究、方法和综述,重点关注微生物组、生物信息、大数据和多组学等前沿交叉学科。目标是发表前10%(IF > 20)的高影响力论文。期刊特色包括中英双语图文、双语视频、可重复分析、图片打磨、60万用户的社交媒体宣传等。2022年2月正式创刊!相继被Google Scholar、PubMed、SCIE、ESI、DOAJ、Scopus等数据库收录!2025年6月影响因子33.2,中科院分区生物学1区Top,位列全球SCI期刊前千分之三(65/22249),微生物学科2/163,仅低于Nature Reviews,学科研究类期刊全球第一,中国大陆5/585!
“iMetaOmics” 是“iMeta” 子刊,主编由中国科学院北京生命科学研究院赵方庆研究员和香港中文大学于君教授担任,目标是成为影响因子大于10的高水平综合期刊,欢迎投稿!
"iMetaMed" 是“iMeta” 子刊,专注于医学、健康和生物技术领域,目标是成为影响因子大于15的医学综合类期刊,欢迎投稿!
iMeta主页:
http://www.imeta.science
姊妹刊iMetaOmics主页:
http://www.imeta.science/imetaomics/
出版社iMeta主页:
https://onlinelibrary.wiley.com/journal/2770596x
出版社iMetaOmics主页:
https://onlinelibrary.wiley.com/journal/29969514
出版社iMetaMed主页:
https://onlinelibrary.wiley.com/journal/3066988x
iMeta投稿:
https://wiley.atyponrex.com/journal/IMT2
iMetaOmics投稿:
https://wiley.atyponrex.com/journal/IMO2
iMetaMed投稿:
https://wiley.atyponrex.com/submission/dashboard?siteName=IMM3
邮箱:
office@imeta.science

















 43
43

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









