









学徒作业1
看懂文章:https://www.jci.org/articles/view/96060/figure/1 看其C子图里面的TRAF4基因在4个数据集的表达量,画出更漂亮的boxplot。
提示:需要看完文章,了解作者所引用的数据并且下载对应的数据集,提取TRAF4基因对应的探针的表达量,根据对应的分组信息画boxplot。
-
2010-cancer cell MSKCC GSE21032 ProstateCancer Genomics Data Portalat http://cbio.mskcc.org/prostate-portal
-
2005-cancer cell GSE3325 Affymetrix U133 2.0 Plus arrays
-
2007-BMCCancer. GSE6919
-
2012- Nature. GEO(GSE35988).
我选择第一个数据集进行分析,以下是原图:

1:更好看的箱线/小提琴/点图绘制
一、数据集下载(仅展示其中一个数据集的分析流程):
首先我在GEO数据库中搜索了GSE21032这个数据集,然而我发现这个数据集通过芯片流程下载可能比较困难,样本数有743个。因此我直接在网站上搜索这个数据集,最后发现这个数据集存在于cbioprotal网站上。

网站上直接搜索

进入cbioportal官网
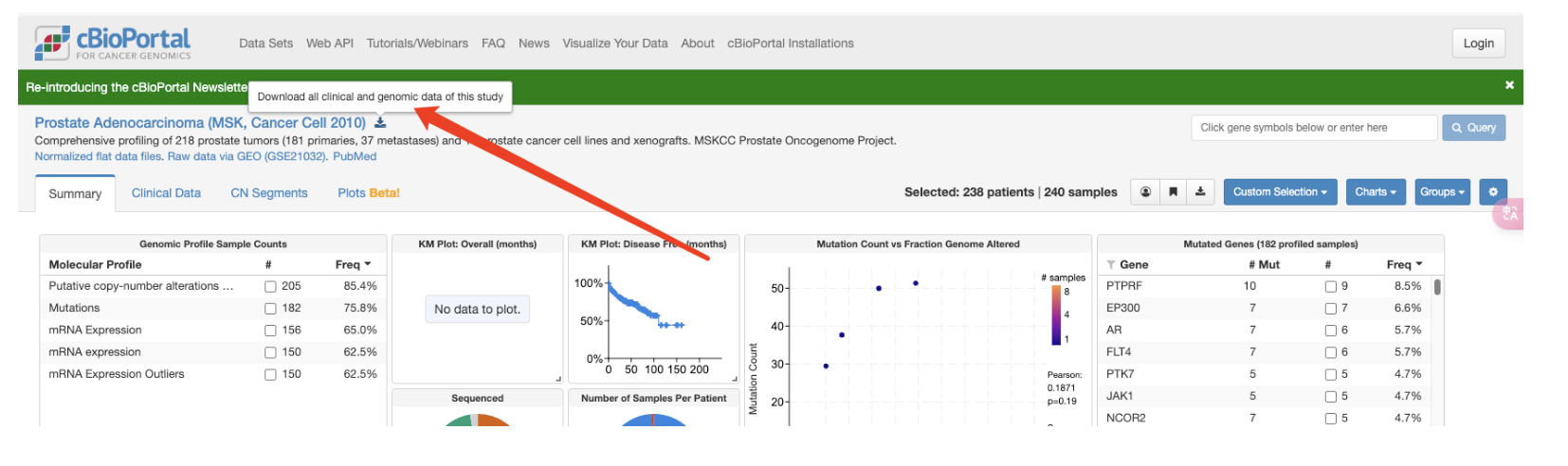
在左上角的地方可以下载到这个数据集的原始数据
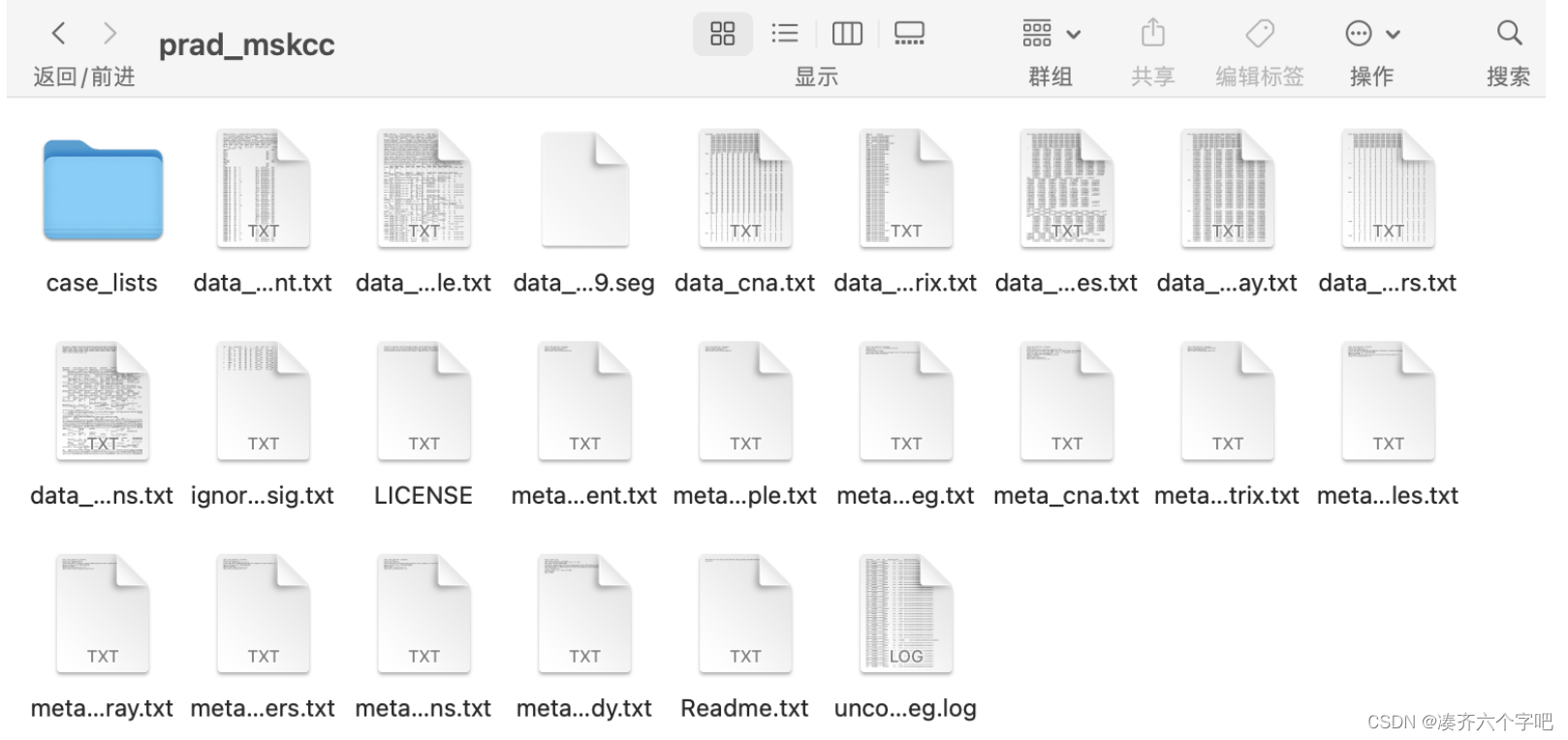
下载之后可以得到这么多的数据。我们需要其中的临床数据和表达矩阵数据。
二、开始分析 1.加载数据
rm(list = ls())
exp <- read.table("data_mrna_agilent_microarray.txt",header = T,check.names = T)
pd <- read.delim("data_clinical_sample.txt",header = T,check.names = T,comment.char = "#")
#值得一提的是,comment.char参数可以把以#号开头的内容去除
2.提取表达矩阵并观察数据情况
#这个数据直接使用的基因ID,所以直接加载人基因数据库
library(dplyr)
library(stringr)
library(org.Hs.eg.db)
ids <- toTable(org.Hs.egSYMBOL)
#接下来就是将exp数据中的行名与ids文件中的基因名按照ID相对应并转换
exp = as.data.frame(exp)
exp = mutate(exp,gene_id = exp$Entrez_Gene_Id)
#ids去重复,按照symbol列中的数据
ids = distinct(ids,symbol,.keep_all = T)
#规范数据格式
exp$gene_id <- as.character(exp$gene_id)
ids$gene_id = as.character(ids$gene_id)
#根据exp中的gene_id进行合并
exp = inner_join(exp,ids,by="gene_id")
nrow(exp)
exp <- exp[,-c(1,158)]
exp = distinct(exp,symbol,.keep_all = T)
rownames(exp) <- exp$symbol
exp <- exp[,-c(157)]
#表达矩阵exp
dim(exp)
range(exp)#看数据范围决定是否需要log,是否有负值,异常值
#exp = log2(exp+1) #需要log才log
boxplot(exp,las = 2) #看是否有异常样本
#如果数据非常不齐,则需要归一化,函数如下
#exp = limma::normalizeBetweenArrays(exp)
#boxplot(exp,las = 2)
3.提取临床信息及排序
rownames(pd) <- pd$SAMPLE_ID
#让exp列名与pd的行名顺序完全一致
p = identical(rownames(pd),colnames(exp));p
if(!p) {
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
}
4.保存数据
save(pd,exp,file = "step1output.Rdata")
三、数据分组及处理
1.加载step1中的数据
rm(list = ls())
load(file = "step1output.Rdata")
2.提取想要的数据-临床数据/表达矩阵
#提取想要的数据
#把前列腺癌细胞系的数据剔除,留下前列腺正常细胞系但数量只有1个
#其实分析到这里的时候我有点犹豫,因为这个数据集在文章中展示是展示了Normal组数据而且绘制了箱线图
#所以我很好奇文章中的正常数据是从哪里得来的
#但不妨碍我后续分析及绘图
library(stringr)
pd = pd[-c(1:5),]
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
identical(rownames(pd),colnames(exp))
3.设置分组
# Group----
library(stringr)
#重新设置Group分组
pd$Group <- ifelse(pd$SAMPLE_TYPE == "Primary","Primary",
ifelse(pd$SAMPLE_TYPE == "Metastasis","Metastasis","Normal"))
#需要把Group转换成因子,并设置参考水平,指定levels,对照组在前,处理组在后
Group = factor(pd$Group,levels = c("Normal","Primary","Metastasis"))
Group
4.保存数据
save(exp,Group,pd,file = "step2output.Rdata")
四、统计分析及绘图
这里我把常用的数据情况及对应的统计方法做了展示。
所以很多函数只是展示在这里,需要先按照正常分析流程需要对每一分组的数据进行正态性检验及方差齐性检验,然后再选择统计检验方法。
该研究来一共有三组(Normal,primary,metastases),需要两两比较,已知在Normal组中只有一个数据,所以有两组数据一定是需要采用非参数检验的,我这边使用了wilcox.test
1.加载数据和R包
rm(list = ls())
load("step2output.Rdata")
2.提取想要的数据
exp_t <- as.data.frame(t(exp))
pd <- cbind(pd,exp_t$TRAF4)
3.正态性检验-对每一组数据进行正态性检验
#检验数据是否满足正态分布的常见方法有:
# Shapiro-Wilk检验,Kolmogorov-Smirnov检验,Anderson-Darling检验,Lilliefors检验,Jarque-Bera检验,D'Agostino's K-squared检验
#最常用以下两种
#Shapiro-Wilk 正态性检验:最常用的正态性检验方法之一。适用于小样本数据。
shapiro.test(pd$`exp_t$TRAF4`)
#Kolmogorov-Smirnov 正态性检验:这是一种基于经验分布函数的检验方法,适用于大样本数据。
ks.test(pd$`exp_t$TRAF4`, "pnorm", mean(pd$`exp_t$TRAF4`), sd(pd$`exp_t$TRAF4`))
#看p值,若满足p-value>0.05,则数据满足正态分布,若不满足则用非参数检验。
#密度直方图
hist(dt$Sepal.Length, main="Histogram for frequency") #默认是probability = F, 频数直方图
hist(dt$Sepal.Length, main="Histogram for density", probability = T) #probability = T,
#QQ图(Quantile-Quantile Plot):这是一种图形方法,可以直观地检查数据是否接近正态分布。
#在QQ图中,如果数据点紧密地遵循参考线(45度线),则数据接近正态分布
qqnorm(pd$`exp_t$TRAF4`)
qqline(pd$`exp_t$TRAF4`)
#一般bartlett.test检查和QQ图用得比较多
#第二种QQ图
library(car)
qqPlot(lm(pd$`exp_t$TRAF4` ~ Group, data=pd),
simulate=TRUE,
main="Q-Q Plot",
lables=FALSE)
abline(0, 1, col = "blue", lwd = 2)
3.方差齐性检验
#方差性(方差齐性)的检验,常用的方法是:
# Bartlett's Test
bartlett.test(`exp_t$TRAF4` ~ Group, data = pd)
# Levene's Test
leveneTest(value ~ group, data = df)
4.若两样本且满足正态+方差齐-采用t.test检验
#t.test检验
result <- t.test(pd$`exp_t$TRAF4` ~ Group, data = pd)
#p值小于0.05,则存在差异。若大于0.05,则无差异。
print(result)
5.若两样本且不满足正态和方差齐-采用Wilcoxon 秩和检验(Mann-Whitney U 检验)
# Wilcoxon 秩和检验
result <- wilcox.test(pd$`exp_t$TRAF4` ~ Group, data = pd)
#p值小于0.05,则存在差异。若大于0.05,则无差异。
print(result)
6.若多样本且满足正态或/和方差齐-单因素方差分析(One-Way ANOVA)
# One-Way ANOVA 检验
result <- wilcox.test(pd$`exp_t$TRAF4` ~ Group, data = pd)
#p值小于0.05,则存在差异。若大于0.05,则无差异。
print(result)
7.若多样本且不满足正态或/和方差齐-Kruskal-Wallis 检验(Mann-Whitney U 检验的拓展)
# One-Way ANOVA 检验
result <- kruskal.test(pd$`exp_t$TRAF4` ~ Group, data = pd)
#p值小于0.05,则存在差异。若大于0.05,则无差异。
print(result)
#Kruskal-Wallis rank sum test
#data: pd$`exp_t$TRAF4` by Group
#Kruskal-Wallis chi-squared = 8.2106, df = 2, p-value = 0.01649
8.Friedman 检验:用于比较三个或更多相关样本(配对样本)
friedman_result <- friedman.test(value ~ condition | subject, data = df)
9.事后分析(如 Dunn's Test)在 Kruskal-Wallis检验显示显著差异后,确定具体哪些组之间存在差异。
# Dunn's Test
#我们也可以做个事后分析,看看哪些组之间存在差异
#install.packages("FSA")
library(FSA)
dunn_result <- dunnTest(pd$`exp_t$TRAF4` ~ Group, data = pd, method = "bonferroni")
print(dunn_result)
# Comparison Z P.unadj P.adj
#1 Metastasis - Normal -0.4996871 0.617295402 1.00000000
#2 Metastasis - Primary 2.6548001 0.007935542 0.02380662
#3 Normal - Primary 1.1599751 0.246058927 0.73817678
-
绘图
# 绘制小提琴图和显著性标记
library(ggplot2)
library(ggpubr)
png("p.png",width = 1200,height = 1200,res = 300)
ggplot(data=pd,aes(x=pd$Group,y=pd$`exp_t$TRAF4`,
colour = pd$Group))+ #fill参数不要设置,会不好看
geom_violin(#color = 'grey',
alpha = 0.8, #alpha = 0.8 参数控制着小提琴图的透明度。
scale = 'width',#小提琴宽度
#linewidth = 1, #外轮廓粗细
trim = TRUE)+ # trim = TRUE 参数控制着小提琴图的形状。
geom_boxplot(mapping=aes(x=pd$Group,y=pd$`exp_t$TRAF4`,
colour=pd$Group,fill=pd$Group), #箱线图
alpha = 0.5,
size=1.5,
width = 0.3)+
geom_jitter(mapping=aes(x=pd$Group,y=pd$`exp_t$TRAF4`,colour = pd$Group), #散点
alpha = 0.3,size=3)+
scale_fill_manual(limits=c("Normal","Primary","Metastasis"),
values =c("#2B6688","#F1A93B","#A8ACB9"))+
scale_color_manual(limits=c("Normal","Primary","Metastasis"),
values=c("#2B6688","#F1A93B","#A8ACB9"))+ #颜色
geom_signif(mapping=aes(x=pd$Group,y=pd$`exp_t$TRAF4`), # 不同组别的显著性
comparisons = list(c("Normal", "Primary"), # 哪些组进行比较
c("Normal", "Metastasis"),
c("Primary", "Metastasis")),
map_signif_level=T, # T显示显著性,F显示p value
tip_length=c(0,0,0,0,0,0),#把向下的帽子去掉,分组数乘以2
y_position = c(10,10.25,10.5), # 设置显著性线的位置高度
size=1, # 修改线的粗细
textsize = 4, # 修改显著性标记的大小
test = "wilcox.test", # 检验的类型,可以更改
color = "black")+ # 设置显著性线的颜色
theme_bw()+ #设置白色背景
guides(fill = guide_legend(title = "Group"), # 设置填充图例的标题
color = guide_legend(title = "Group"))+ # 设置颜色图例的标题
labs(x="Group",y="TRAF4 expression") # 添加标题,x轴,y轴标签
dev.off()
 修改后的美图
修改后的美图
小结:通过这题学习可以加深我们对选择哪一种统计学工具去分析数据的认识且训练了我们绘制箱线/小提琴/点图的能力。
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员(部分代码来源:生信技能树马拉松和数据挖掘课程)。
注:若对代码有疑惑或者发现有明确错误的,请联系后台(希望多多交流)。更多内容可关注公众号:生信方舟
- END -















 203
203











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









