







学徒作业
理论上,任意疾病或者其它实验设计,都是可以找到多个数据集,它们各自可以独立差异分析。然后,使用下面的统计学方法和工具来进行比较:1.Jaccard相似性指数;2.Pearson相关系数;3. Spearman秩相关系数;4. Venn图;5. Gene Set Enrichment分析(GSEA); 6. 差异基因列表重叠分析; 7. 回归分析。
一、数据集选择: GSE13601 和GSE9844 (随机挑选)
1.GSE13601数据集:芯片数据,内含29例舌癌样本和29例正常对照样本(舌)。
2.GSE9844数据集:芯片数据,内含26例舌癌样本和12例正常对照样本。
二、芯片数据分析标准流程(仅演示一个数据)
1.设置下载时长
rm(list = ls())
#打破下载时间的限制,改前60秒,改后1000w秒
options(timeout = 10000000)
options(scipen = 20)#不要以科学计数法表示
2.传统芯片下载方式
#传统下载方式
library(GEOquery)
eSet = getGEO("GSE13601", destdir = '.', getGPL = F)
#研究一下这个eSet
class(eSet)
length(eSet)
eSet = eSet[[1]]
class(eSet)
3.提取表达矩阵并观察数据情况
#(1)提取表达矩阵exp
exp <- exprs(eSet)
dim(exp)
range(exp)#看数据范围决定是否需要log,是否有负值,异常值
exp = log2(exp+1) #需要log才log
boxplot(exp,las = 2) #看是否有异常样本
#如果数据非常不齐,则需要归一化,函数如下
exp = limma::normalizeBetweenArrays(exp)
boxplot(exp,las = 2)
4.提取临床信息及排序
# 提取临床信息
pd <- pData(eSet)
#让exp列名与pd的行名顺序完全一致
p = identical(rownames(pd),colnames(exp));p
if(!p) {
s = intersect(rownames(pd),colnames(exp))
exp = exp[,s]
pd = pd[s,]
}
5.保存数据
# 提取芯片平台编号,后面要根据它来找探针注释
gpl_number <- eSet@annotation;gpl_number
save(pd,exp,gpl_number,file = "step1output.Rdata")
6.加载step1中的数据
rm(list = ls())
load(file = "step1output.Rdata")
7.设置分组-Group
# Group----
library(stringr)
# 使用字符串处理的函数获取分组
k = str_detect(pd$title,"Normal");table(k)
Group = ifelse(k,"Normal","Tumor")
# 需要把Group转换成因子,并设置参考水平,指定levels,对照组在前,处理组在后
Group = factor(Group,levels = c("Normal","Tumor"))
Group
8.探针注释
# 探针注释的获取-----------------
library(tinyarray)
find_anno(gpl_number) #打出找注释的代码
ids <- AnnoProbe::idmap('GPL8300')
#为exp数据框添加几列
#1.加probe_id列,把行名变成一列
library(dplyr)
exp = as.data.frame(exp)
exp = mutate(exp,probe_id = rownames(exp))
#2.加上探针注释
ids = distinct(ids,symbol,.keep_all = T)
class(ids);class(exp)
ids$probe_id = as.character(ids$probe_id)
exp = inner_join(exp,ids,by="probe_id")
nrow(exp)
exp = select(exp,-"probe_id")
rownames(exp) <- exp$symbol
exp = select(exp,-"symbol")
9.保存数据
save(exp,Group,ids,file = "step2output.Rdata")
10.加载step2中的数据
rm(list = ls())
load(file = "step2output.Rdata")
11.PCA绘制
# 1.PCA 图----
dat=as.data.frame(t(exp))
library(FactoMineR)
library(factoextra)
dat.pca <- PCA(dat, graph = FALSE)
fviz_pca_ind(dat.pca,
geom.ind = "point", # show points only (nbut not "text")
col.ind = Group, # color by groups
palette = c("#00AFBB", "#E7B800"),
addEllipses = TRUE, # Concentration ellipses
legend.title = "Groups"
)
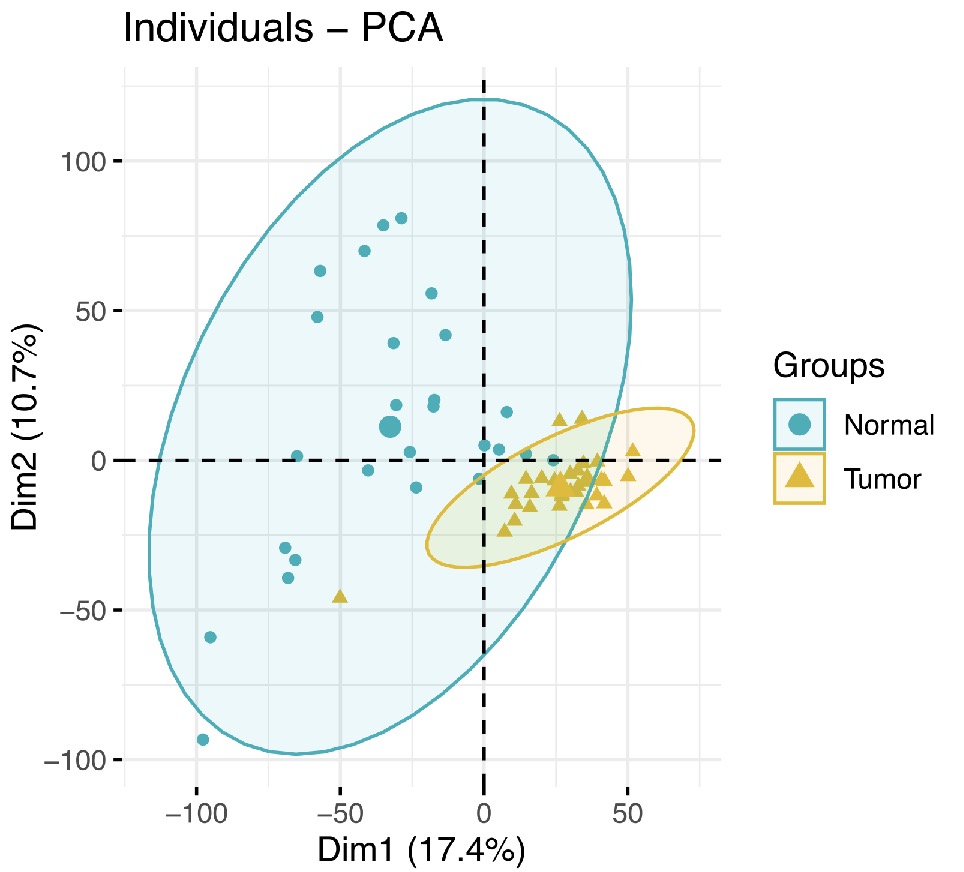
GSE13601-PCA
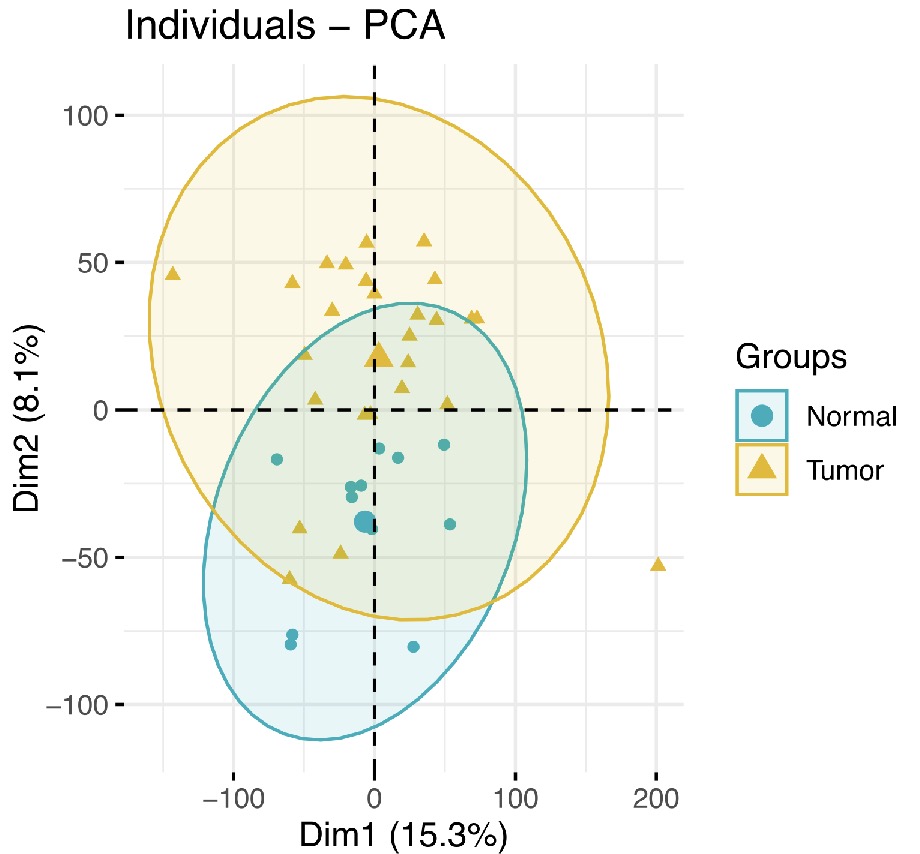
GSE9844-PCA
从两个图来看,这两个数据集中的肿瘤与非肿瘤区分情况都还行。
12.limma差异分析-芯片数据只能用limma
#差异分析
library(limma)
design = model.matrix(~Group)
fit = lmFit(exp,design)
fit = eBayes(fit)
deg = topTable(fit,coef = 2,number = Inf)
13.设置change列(上下调基因)
# 加change列,标记上下调基因
logFC_t = 1 #logFC值我设置了1
p_t = 0.05
k1 = (deg$P.Value < p_t)&(deg$logFC < -logFC_t)
k2 = (deg$P.Value < p_t)&(deg$logFC > logFC_t)
deg = mutate(deg,change = ifelse(k1,"down",ifelse(k2,"up","stable")))
table(deg$change)
GSE13601数据集中上下调基因情况: down(527),stable(7293) ,up(765)
值得一提的是,这个数据集中的基因总数约为8000+,但实际上咱们人类中应该至少存在20000个已知的基因。
GSE9844数据集中上下调基因情况:down(280),stable(19642) ,up(266)
这个数据集中的基因总数就比较正常。
14.差异基因火山图
#火山图
library(ggplot2)
ggplot(data = deg, aes(x = logFC, y = -log10(P.Value))) +
geom_point(alpha=0.4, size=3.5, aes(color=change)) +
scale_color_manual(values=c("blue", "grey","red"))+
geom_vline(xintercept=c(-logFC_t,logFC_t),lty=4,col="black",linewidth=0.8) +
geom_hline(yintercept = -log10(p_t),lty=4,col="black",linewidth=0.8) +
theme_bw()
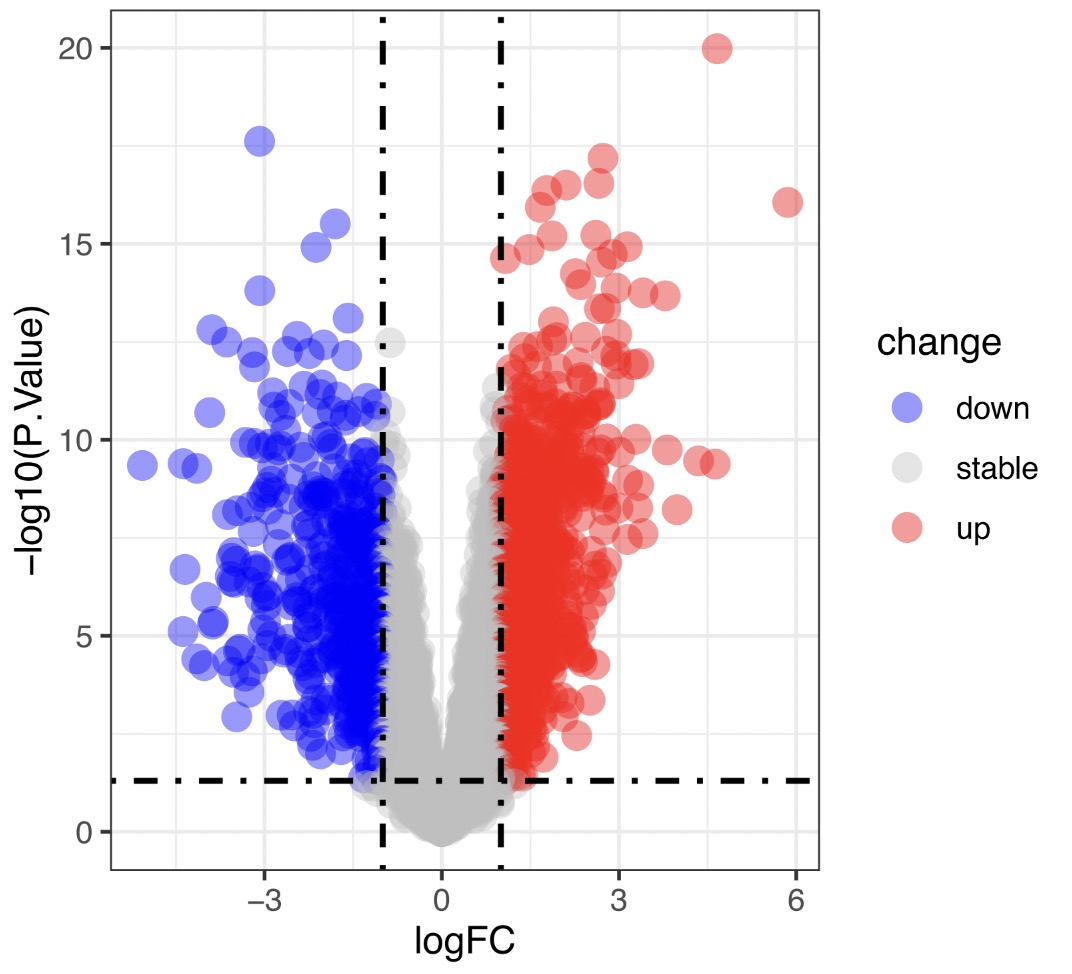
GSE13601-火山图(仅展示一个数据集)
15.保存数据
save(exp,Group,deg,logFC_t,p_t,file = "step4output.Rdata")
小结:我们可以发现,虽然是相同来源的样本,分组情况也相似,设置的分析参数也是一致的,但最后得出的上下调基因数量就是存在差异,这也就如曾老师所提到的存在其他的混杂因素。
致谢:感谢曾老师,小洁老师以及生信技能树团队全体成员(代码来源:生信技能树马拉松和数据挖掘课程)。
注:若对内容有疑惑或者发现有明确错误的,请联系后台(希望多多交流)。更多内容可见公众号:生信方舟。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









