









irGSEA整合了多种基于单个细胞表达等级的富集分析方法(AUCell、UCell、singscore、ssGSEA、JASMINE和Viper),并通过秩聚合算法(robust rank aggregation, RRA)对差异分析的结果进行评估,筛选出在这种几种方法中表现出相似的富集程度的差异基因集。
通俗的来说,这个R包就是整合了6种富集分析方法并对结果再次打分取交集,获取在这6种方法中均表现相似的基因集。
分析步骤-自己数据实践一下
1、irGSEA安装
大多数人只需要这么安装即可,如果需要使用python还可以参考开发者的进阶版。
# install packages from CRAN
cran.packages <- c("aplot", "BiocManager", "data.table", "devtools",
"doParallel", "doRNG", "dplyr", "ggfun", "gghalves",
"ggplot2", "ggplotify", "ggridges", "ggsci", "irlba",
"magrittr", "Matrix", "msigdbr", "pagoda2", "pointr",
"purrr", "RcppML", "readr", "reshape2", "reticulate",
"rlang", "RMTstat", "RobustRankAggreg", "roxygen2",
"Seurat", "SeuratObject", "stringr", "tibble", "tidyr",
"tidyselect", "tidytree", "VAM")
for (i in cran.packages) {
if (!requireNamespace(i, quietly = TRUE)) {
install.packages(i, ask = F, update = F)
}
}
# install packages from Bioconductor
bioconductor.packages <- c("AUCell", "BiocParallel", "ComplexHeatmap",
"decoupleR", "fgsea", "ggtree", "GSEABase",
"GSVA", "Nebulosa", "scde", "singscore",
"SummarizedExperiment", "UCell",
"viper","sparseMatrixStats")
for (i in bioconductor.packages) {
if (!requireNamespace(i, quietly = TRUE)) {
install.packages(i, ask = F, update = F)
}
}
# install packages from Github
if (!requireNamespace("irGSEA", quietly = TRUE)) {
devtools::install_github("chuiqin/irGSEA", force =T)
}
2、check数据
rm(list = ls())
library(irGSEA)
library(Seurat)
library(SeuratData)
library(RcppML)
sc_dataset <- readRDS("./sc_dataset.rds")
# Check
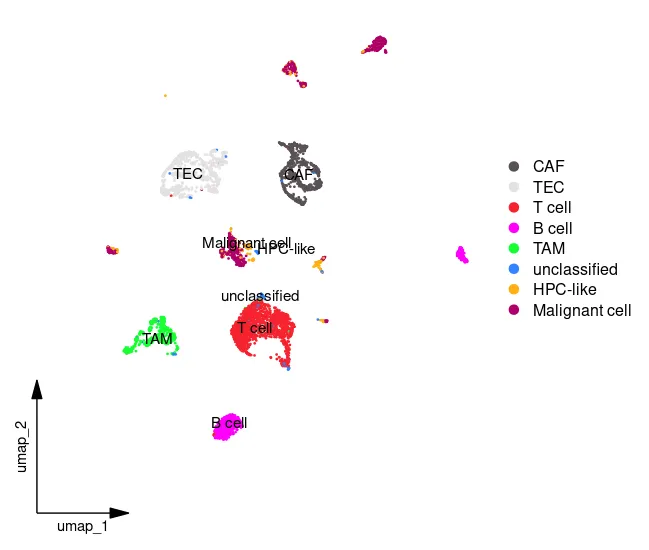
UMAP_celltype <- DimPlot(sc_dataset, reduction ="umap",
group.by="celltype",label = T);UMAP_celltype
Idents(sc_dataset) <- sc_dataset$celltype
scCustomize::DimPlot_scCustom(sc_dataset, figure_plot = TRUE)
3、irGSEA计算富集分数
如果是Seurat V4或者其他的需要参考开发者的教程。
#### Seurat V5对象 ####
sc_dataset <- SeuratObject::UpdateSeuratObject(sc_dataset)
sc_dataset2 <- CreateSeuratObject(counts = CreateAssay5Object(GetAssayData(sc_dataset,
assay = "RNA",
slot="counts")),
meta.data = sc_dataset[[]])
sc_dataset2 <- NormalizeData(sc_dataset2)
sc_dataset2 <- irGSEA.score(object = sc_dataset2, assay = "RNA",
slot = "data", seeds = 123,
#ncores = 1,
min.cells = 3, min.feature = 0,
custom = F, geneset = NULL, msigdb = T,
species = "Homo sapiens",
category = "H",
subcategory = NULL,
geneid = "symbol",
method = c("AUCell","UCell","singscore",
"ssgsea", "JASMINE", "viper"),
aucell.MaxRank = NULL,
ucell.MaxRank = NULL,
kcdf = 'Gaussian')
min.cells = 3: 指定最少需要多少个细胞表达某个基因才能将其包括在分析中。设置为3,意味着基因必须在至少3个细胞中表达才能被考虑。
min.feature = 0: 指定最少特征数(基因或其他特征)阈值,这里设置为0,表示不进行特征过滤。
custom = F: 指定是否使用自定义基因集。这里设置为 False,表示不使用自定义基因集。
geneset = NULL: 如果 custom = TRUE,则需要提供自定义的基因集。这里设置为 NULL,表示不使用自定义基因集。
msigdb = T: 指定是否使用 MSigDB(Molecular Signatures Database)中的基因集。T 表示使用。
species = "Homo sapiens": 指定要分析的物种,这里选择的是人类(Homo sapiens)。
category = "H": 指定基因集的类别。在 MSigDB 中,“H” 表示 hallmark 基因集,这是一组预先定义的基因集,用于捕捉广泛生物学过程中的特征。
subcategory = NULL: 子类别的指定,这里为 NULL,表示不指定任何子类别。
geneid = "symbol": 指定基因ID类型,这里使用的是基因符号(symbol),即基因的标准命名。
method = c("AUCell","UCell","singscore","ssgsea", "JASMINE", "viper"): 指定要使用的富集方法列表。
aucell.MaxRank = NULL 和 ucell.MaxRank = NULL: 这些参数用于设置 AUCell 和 UCell 方法的最大排名,这里设置为 NULL,表示使用默认值。
kcdf = 'Gaussian': 设置核密度估计的类型,这里使用高斯(Gaussian)分布。
研究者还贴心的为使用者内置了msigdbr包进行打分,可以通过修改category = "H", subcategory = NULL,参数进行比如Hallmark、KEGG,GO等评分。
# 整合差异基因集
# 如果报错,考虑加句代码options(future.globals.maxSize = 100000 * 1024^5)
result.dge <- irGSEA.integrate(object = sc_dataset2,
group.by = "celltype",
method = c("AUCell","UCell","singscore",
"ssgsea", "JASMINE", "viper"))
# 查看RRA识别的在多种打分方法中都普遍认可的差异基因集
geneset.show <- result.dge$RRA %>%
dplyr::filter(pvalue <= 0.05) %>%
dplyr::pull(Name) %>% unique(.)
4、可视化
热图
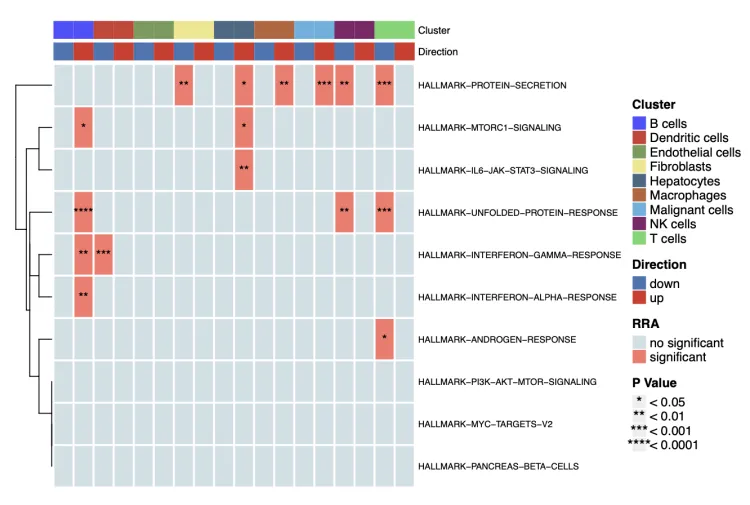
heatmap.plot <- irGSEA.heatmap(object = result.dge,
method = "RRA",
top = 10,
show.geneset = NULL)
heatmap.plot
top参数可以修改通路的数量,这次设置了10。
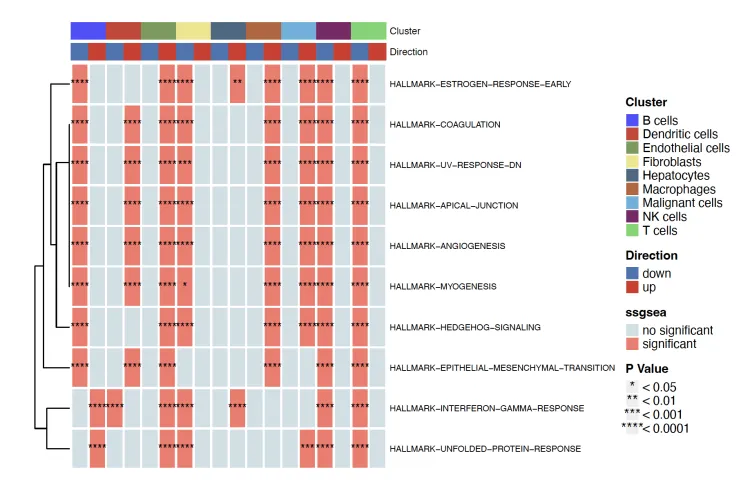
heatmap.plot <- irGSEA.heatmap(object = result.dge,
method = "ssgsea”
top = 10,
show.geneset = NULL)
heatmap.plot
method = "ssgsea", #从'RRA"换成其他的单独分析方法,这里尝试使用了“ssgsea”。
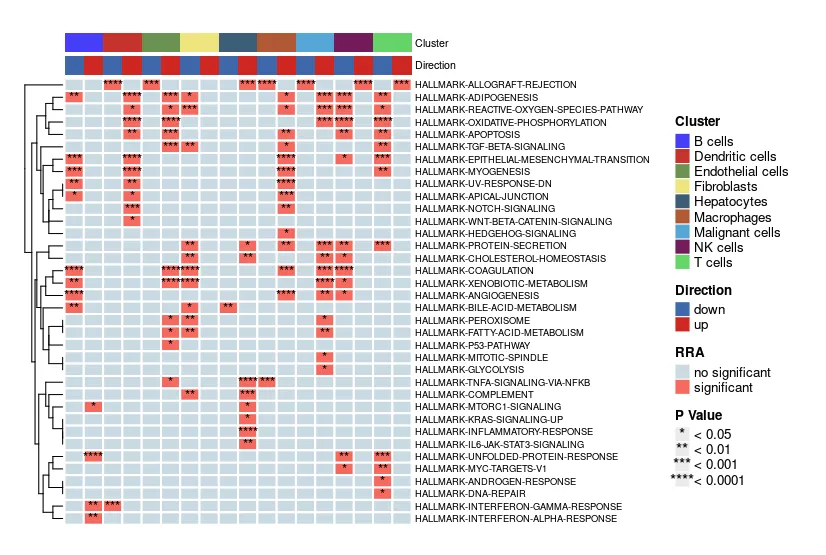
heatmap.plot <- irGSEA.heatmap(object = result.dge,
method = "RRA", #从'RRA"换成“ssgsea”
top = 10,
show.geneset = geneset.show)
heatmap.plot
在show.geneset后边使用geneset.show。这种方式是可以指定在当前method下展示具有统计学意义的通路。此时就算是设置top10也没有用。
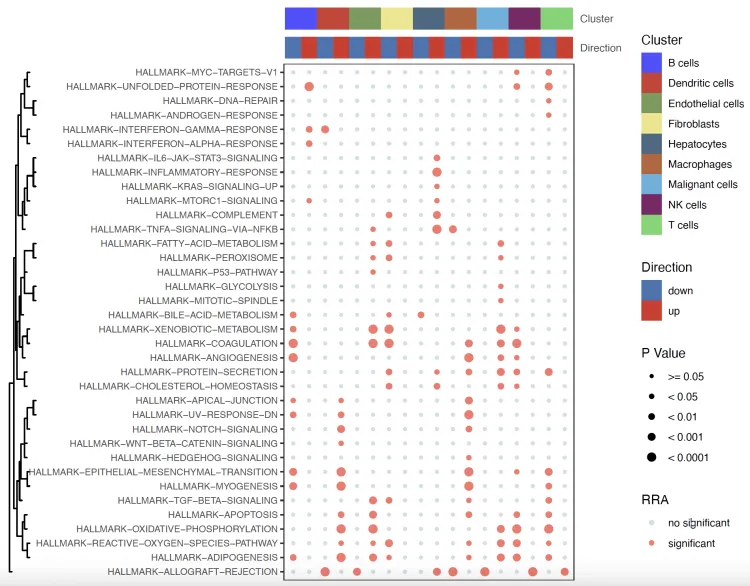
气泡图
bubble.plot <- irGSEA.bubble(object = result.dge,
method = "RRA",
top = 10,
show.geneset = geneset.show)
bubble.plot
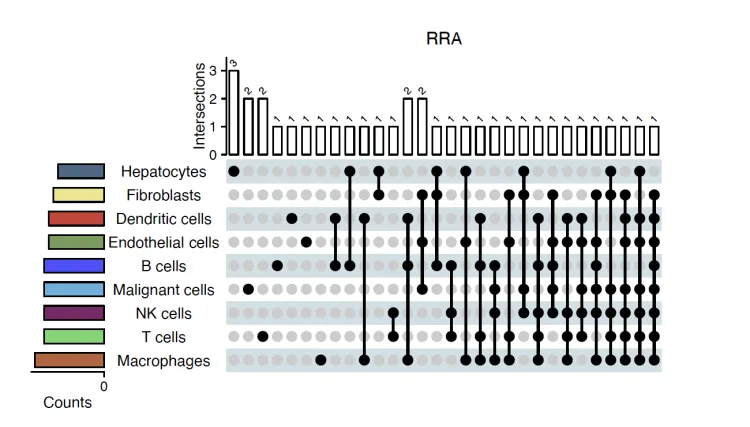
Upset图
upset.plot <- irGSEA.upset(object = result.dge,
method = "RRA")
upset.plot
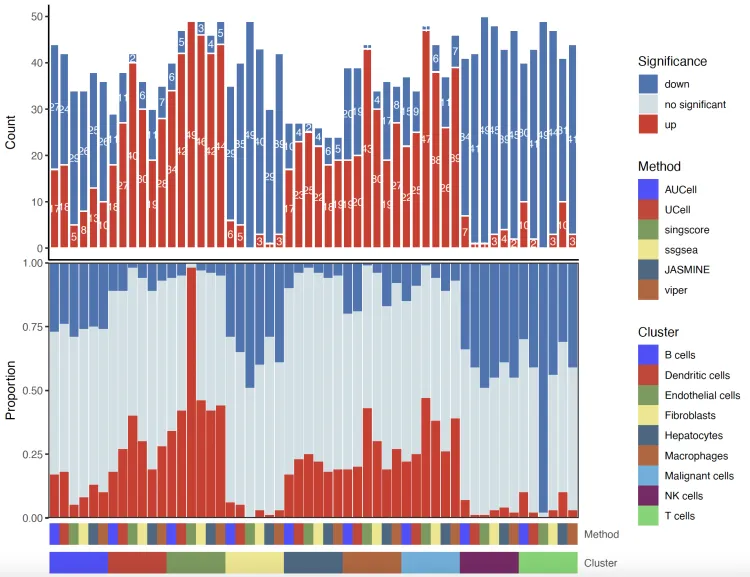
堆叠条形图
barplot.plot <- irGSEA.barplot(object = result.dge,
method = c("AUCell","UCell","singscore",
"ssgsea", "JASMINE", "viper"))
barplot.plot
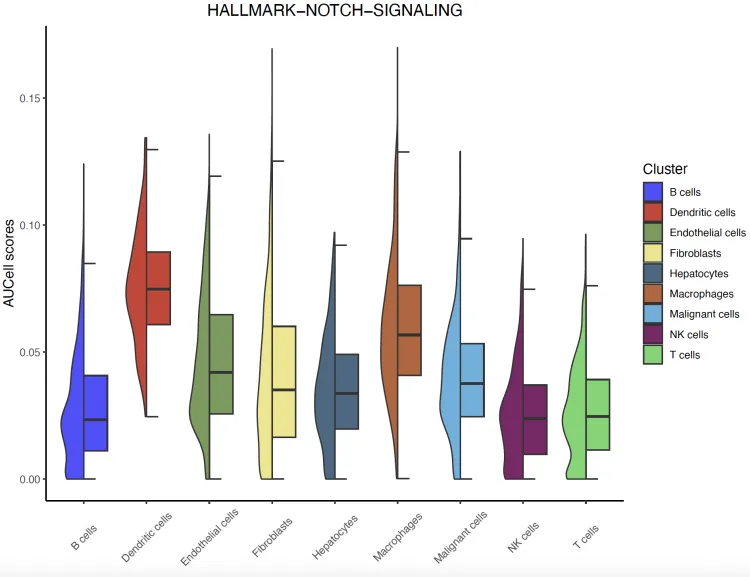
半小提琴图
Idents(sc_dataset2) <- sc_dataset2$celltype
halfvlnplot <- irGSEA.halfvlnplot(object = sc_dataset2,
method = "AUCell",
show.geneset = "HALLMARK-NOTCH-SIGNALING")
halfvlnplot
vlnplot <- irGSEA.vlnplot(object = sc_dataset2,
method = c("AUCell", "UCell",
"singscore", "ssgsea",
"JASMINE", "viper"),
show.geneset = "HALLMARK-INFLAMMATORY-RESPONSE")
vlnplot
其他的图就不展示了,大同小异的。
#山峦图
ridgeplot <- irGSEA.ridgeplot(object = sc_dataset2,
method = "AUCell",
show.geneset = "HALLMARK-NOTCH-SIGNALING")
ridgeplot
#密度热图
densityheatmap <- irGSEA.densityheatmap(object = sc_dataset2,
method = "AUCell",
show.geneset = "HALLMARK-NOTCH-SIGNALING")
densityheatmap
总的来说这个R包还是很方便的,而且多种富集方式的交集可以给后续分析带来更坚实有力的证据支撑。
参考资料:
1、irGSEA:https://github.com/chuiqin/irGSEA
2、范垂钦_92be 老师/生信技能树:https://mp.weixin.qq.com/s/VI4ISO6y5_rt8Yy_VnIR-w
致谢:感谢曾老师以及生信技能树团队全体成员。
注:若对内容有疑惑或者有发现明确错误的朋友,请联系后台(欢迎交流)。更多内容可关注公众号:生信方舟
- END -
















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









