






ReaxFF力场的发展弥合量子化学方法和经典非反应性分子动力学力场之间的差距,ReaxFF力场是一个键级相互作用模型,这一方法突破了传统力场中原子间固定化学键连接的局限,引入了键级这一动态概念来描述原子间的化学性质。随着模拟过程中化学键的断裂与生成,原子间的性质也在不断变化,从而实现了对化学反应动态的过程更真实模拟。其中ReaxFF势函数将总势能细分为多个组成部分,包含了键相关作用项和非键相关作用项总体构成如式1所示:
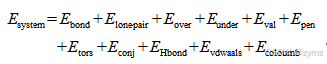
式中,体系的总能量(Esystem)是根据各种能量贡献计算的,包括键能(Ebond)、孤对电子能(Elone-pair)、过配位能(Eover)、欠配位能量(Eunder)、键角能(Eval)、二面角能(Etors),以及非键相互作用如范德华作用能(Evdwaals)和库仑作用能(Ecoloumb)。ReaxFF力场文件定义了分子间和分子内的相互作用,包括基本参数、原子参数、键参数、键角参数、二面角参数、非对角作用项以及氢键参数。在ReaxFF的参数体系中,键级(BO)和键能是核心要素,它们通过原子间的距离rij来判定。具体来说,ReaxFF假定了两个原子之间的相互作用可以通过它们之间的距离rij来确定键的类型,如单键(σ键)、双键(π键)和三键(ππ键)。这些不同类型的键在ReaxFF中通过不同的键级来表示,而每个键级都对应着一定的键能。所有键级的总和构成了总键级,它综合反映了原子间相互作用的强度和类型。键级公式如式2所示:
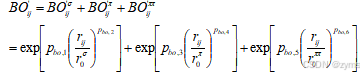
式中,键级是通过一系列参数来描述的,其中Pbo1和Pbo2代表σ键的系数,Pbo3和Pbo4则代表π键的系数,而Pbo5和Pbo6则对应于ππ键系数。这些参数与分子势能面上原子间距离rij的变化密切相关。通过分析键级的变化趋势,理解分子间成键的性质。当原子间的距离rij逐渐减小时,键级会趋近于1,这反映了原子间相互作用逐渐增强,趋向于形成稳定的化学键,相反,当原子间的距离增加时,键级会趋近于0,这表明原子间的相互作用减弱,化学键趋于断裂。通过计算不同键级的成键趋势,可获得ReaxFF力场中不同键的成键距离信息。
在获取键级信息之后,能够进一步计算键级的总体势能。这些总体势能的计算通常基于一系列精细的参数方程,这些参数在优化过程也起一定作用,具体由公式3给出:
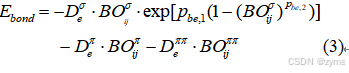
式中,![]()
分别表示σ键、π键和ππ键的阱深。而pbe1和pbe2分别描述的是势阱宽度。van Duin等开发的SOPPI单参数优化算法。虽然SOPPI算法通过逐个训练参数直至满足条件,但这种方法存在陷入局部最小点的风险。除了SOPPI外,还有遗传算法(GA)、模拟退火法(SA)和粒子群优化(PSO)等优化算法可供选择。
关于优化算法安装,请联系本人,留言和私信都可以。

















 5218
5218

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









