






在使用R,Linux,conda虚拟环境里面安装一些R包,总是会遇到一些报错,在这里总结一下,当作学习笔记。
在linux服务器上用conda创建新环境,在新环境里面安装一些R包和python库
在 conda 里面创建新环境并在其中安装包有很多好处,特别是在数据科学、机器学习和软件开发中:
1. 依赖管理
不同的项目可能需要不同版本的包和库。如果所有项目都使用相同的环境,可能会出现包版本冲突的问题。通过创建独立的环境,你可以确保每个项目使用正确版本的依赖库。
2. 隔离性
每个环境是相互独立的,意味着一个环境中的更改不会影响到其他环境。这种隔离性可以防止由于更改某个包版本而导致其他项目出现问题。
3. 可移植性
你可以创建一个包含所有依赖的环境,并与其他人分享。这样可以确保他们在相同的环境下运行代码,从而避免因环境不同导致的运行错误。
4. 重现性
在科学计算和数据分析中,重现性非常重要。通过使用 conda 环境,你可以确保在不同时间、不同设备上运行相同的代码会产生相同的结果。
5. 简化管理
conda 提供了简单的命令行工具来创建、激活、停用和删除环境。这使得环境管理变得非常容易,尤其是对需要同时处理多个项目的开发人员来说。
实际例子
场景 1:不同的Python版本
假设你有两个项目,一个使用Python 2.7,另一个使用Python 3.8。在同一系统上同时管理这两个项目的依赖可能会很麻烦。通过创建两个独立的环境,你可以轻松地在它们之间切换:
# 为项目1创建Python 2.7环境
conda create --name project1 python=2.7
# 为项目2创建Python 3.8环境
conda create --name project2 python=3.8
使用 conda create 命令:
使用 conda create 命令来创建一个新环境。你需要指定环境的名称
conda create --name myenv #创建一个名称叫myenv的环境
conda activate myenv #创建好环境后,激活环境
#查看已创建的所有环境列表,下面两句命令都可以
conda env list
conda info --envs
conda deactivate #停用当前环境安装R包有哪些途径、方法?每个途径有哪些优点和缺点?
在激活的 conda 环境中安装 R 包有几种方法,主要包括使用 conda 安装、使用 R 自带的 install.packages 函数以及使用 Bioconductor 安装。如果某些包在一个源中找不到,可以尝试其他源。
1. 使用 conda 安装 R 包
conda 本身支持安装很多常用的 R 包,这种方式简单且容易管理依赖关系。
优点
-
依赖管理:
conda自动处理和安装包所需的所有依赖项这样可以避免手动解决包与包之间的冲突和兼容性问题。 -
环境隔离:不同项目可以在不同环境中运行,互不影响。
-
简便性:
conda命令行工具使用简单,可以快速安装和管理包。 -
跨平台支持:
conda支持 Windows、macOS 和 Linux,可以在任何平台上使用相同的命令安装包。确保在不同操作系统上有一致的体验。
缺点
-
包的可用性:并非所有的 R 包都可以通过
conda获得,有些包可能在conda频道中不存在 -
频道管理:有时需要配置多个
conda频道才能找到所有需要的包,这可能会导致冲突或兼容性问题。什么是频道?在conda中,频道(channel)是存储和分发包的源。每个频道可以包含多个包及其不同版本。这允许用户从不同的来源获取需要的包conda使用这些频道来查找、下载和安装包。 -
混合安装问题:如果你在同一环境中混合使用
conda和install.packages()安装包,可能会导致依赖冲突或包管理复杂化。 -
包更新:
conda中的包更新频率可能比 CRAN 或 Bioconductor 要慢。
常见的频道
-
默认频道:
conda默认的频道是 Anaconda 官方提供的,包含了大量常见的包和工具。 -
conda-forge:这是一个由社区维护的频道,提供了大量的最新包和包版本。许多用户选择使用
conda-forge因为它包含了更多更新更快的包。 -
bioconda:这是专门用于生物信息学的频道,包含了大量生物信息学工具和包。
-
其他专用频道:有些组织或个人会创建自己的频道,提供特定领域或项目的包。
举个例子:
conda install -c conda-forge r-ggplot2
#conda install:安装命令。
#-c conda-forge:指定使用 conda-forge 频道。
#r-ggplot2:要安装的包名。
#安装特定版本的 R 包,可以在包名称后面加上版本号。例如,安装 ggplot2 的 3.3.3 版本:
conda install -c conda-forge r-ggplot2=3.3.3
conda config --add channels conda-forge
conda config --set channel_priority strict
#这个命令将 conda-forge 添加到频道列表,并设置严格的频道优先级,确保优先从 conda-forge 安装包。推荐使用 conda 的情况
- 当你的项目依赖多个不同语言的包(如 R 和 Python)。
- 当你需要为不同的项目创建独立的开发环境。
- 当你在多个操作系统上开发并希望有一致的安装体验。
- 当你需要自动管理复杂的依赖关系。
使用 R 的 install.packages 函数
在 R 环境中,你可以直接使用 install.packages 函数从 CRAN 安装 R 包
install.packages("packagename") #不能指定包的版本
3. 使用 Bioconductor 安装 R 包
Bioconductor 是一个专门用于生物信息学的 R 包集合。可以使用 Bioconductor 的 BiocManager 来安装包
要在使用 BiocManager::install() 安装包时指定 BiocManager 的版本,你需要首先确保你已经安装了你希望使用的 BiocManager 版本
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager") #安装BiocManager
BiocManager::install(version = "3.19") #安装特定版本BiocManager
BiocManager::version() #查看版本
BiocManager::install("GenomicFeatures") #不能指定安装包的版本,但是可以指定BiocManager的版本
BiocManager::install("GenomicFeatures",version="3.19") #指定BiocManager的版本,用3.19版本的指定BiocManager的版安装GenomicFeaturesBiocManager 安装的优缺点
优点:
-
依赖管理:
BiocManager会自动处理 Bioconductor 包及其所有依赖项,确保包的兼容性。 -
版本控制:你可以指定 Bioconductor 版本,确保在特定版本的 Bioconductor 中安装和使用包。
-
社区支持:Bioconductor 是生物信息学和计算生物学领域的一个主要包管理平台,提供了大量的专用工具和包,受到广泛的社区支持。
-
兼容性测试:Bioconductor 包通常经过严格的测试,确保它们在一起工作时的兼容性和稳定性。
缺点:
-
安装速度:由于一些包依赖于大型数据或需要编译,安装 Bioconductor 包可能比 CRAN 包要慢。
-
平台依赖:有些 Bioconductor 包可能对特定的平台(如 Windows、macOS、Linux)有依赖性问题,需要额外的配置。
-
更新频率:Bioconductor 通常每半年发布一次新的版本,这可能导致一些包版本的延迟更新。如果你需要最新的功能和修复,有时需要等待。
总结
BiocManager 是管理 Bioconductor 包的推荐工具,因为它可以自动处理依赖和版本兼容性问题。你可以通过先安装特定版本的 BiocManager 来指定使用哪个版本。尽管 BiocManager 有一些缺点,比如安装速度和平台依赖性,但其优势在于强大的依赖管理和广泛的社区支持,特别适合处理生物信息学和计算生物学任务
使用 devtools 从 GitHub 安装
有时候你需要从 GitHub 安装开发版本的 R 包,可以使用 devtools 包来安装
devtools 包是 R 语言中的一个工具包,旨在简化 R 包的开发和管理。它提供了一系列功能来帮助开发者创建、测试、文档化和发布 R 包。devtools 还支持从 GitHub 等在线代码库安装 R 包。
if (!requireNamespace("devtools", quietly = TRUE))
install.packages("devtools")
devtools::install_github("hhoeflin/hdf5r")
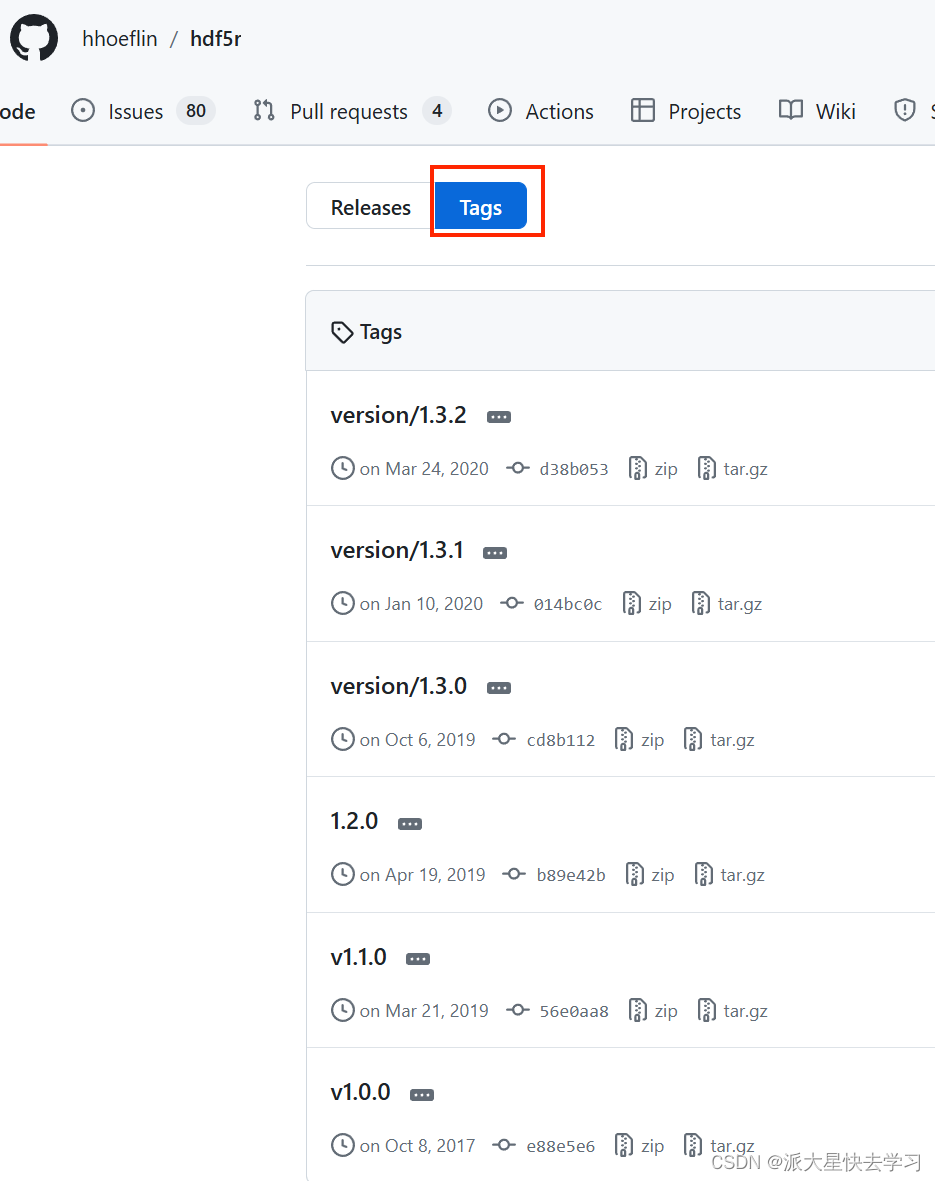
可以看到在GitHub上面发布了好几个版本,当我们想在GitHub上面下载特定版本的时候要指定GitHub上有的版本
devtools::install_github("cran/hdf5r@v1.3.2")
假设你找到的提交哈希是 abc1234,可以这样安装:
devtools::install_github("cran/hdf5r@abc1234")
检查 GitHub 仓库的版本标签或提交哈希
- 访问 GitHub 仓库:打开浏览器,访问
hdf5r的 GitHub 仓库,例如https://github.com/hhoeflin/hdf5r。 - 查找版本标签:在仓库页面上,点击 "Tags" 或 "Releases" 以查找你需要的版本标签。如果存在
v1.3.2版本标签,可以直接使用它。 - 查找提交哈希:如果没有版本标签,可以浏览仓库的提交历史(点击 "Commits")来找到对应版本的提交哈希。
总结:
devtools 是一个强大的工具包,简化了 R 包的开发和管理。使用 devtools 从 GitHub 安装包,可以获取最新的功能和修复,但也有可能遇到稳定性和依赖性问题。适用于需要最新功能、测试和开发、使用实验性功能或参与开源项目的情况。
手动下载并安装
- 手动下载特定版本的包源代码:
你可以从 CRAN Archive 页面(如 https://cran.r-project.org/src/contrib/Archive/ggplot2/)下载所需版本的包。或者是Bioconductor 或 GitHub 等网站下载你想要的包的源代码,通常是 .tar.gz 文件。
2.使用 install.packages() 安装已下载的包:
install.packages("path/to/downloaded/package.tar.gz",
repos = NULL, type = "source") #路径是下载下来的包存放的位置
install.packages():R 用于安装包的函数。"path/to/downloaded/package.tar.gz":这是你下载的 R 包源代码文件的路径。将其替换为实际的文件路径。repos = NULL:告诉 R 不从 CRAN 或其他在线仓库安装包,而是从本地文件安装。type = "source":指定安装包的类型为源代码(而不是二进制包)
总结
手动安装 R 包提供了灵活性,但也有很多潜在的复杂性和缺点,特别是在处理依赖项和更新时。一般情况下,推荐使用 install.packages() 和 BiocManager::install() 等自动处理依赖和更新的工具,除非你有特定需求或限制。
有些时候安装包会报错,可能是因为环境问题,重启R,重新安装可以解决问题,有时候是依赖包没有安装好,这时候,先把依赖包安装好,然后再安装需要的包就可以了。















 976
976

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









