






最近发现2022年出了一个可以做单细胞代谢定量的R包scMetabolism,想着用一下试试。
scMetabolism文章链接:Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level - PubMed (nih.gov)

GitHub链接:
wu-yc/scMetabolism: Quantifying metabolism activity at the single-cell resolution (github.com)
我先根据GitHub的链接,安装依赖包
install.packages(c("devtools", "data.table", "wesanderson", "Seurat", "devtools", "AUCell", "GSEABase", "GSVA", "ggplot2","rsvd"))
devtools::install_github("YosefLab/VISION@v2.1.0") #Please note that the version would be v2.1.0devtools::install_github("wu-yc/scMetabolism")安装的过程中出现了问题说是GSVA,VISION@v2.1.0安装不上

师兄说VISION@v2.1.0安装不上是需要安装一个动态库,先在服务器上安装动态库再安装VISION@v2.1.0。
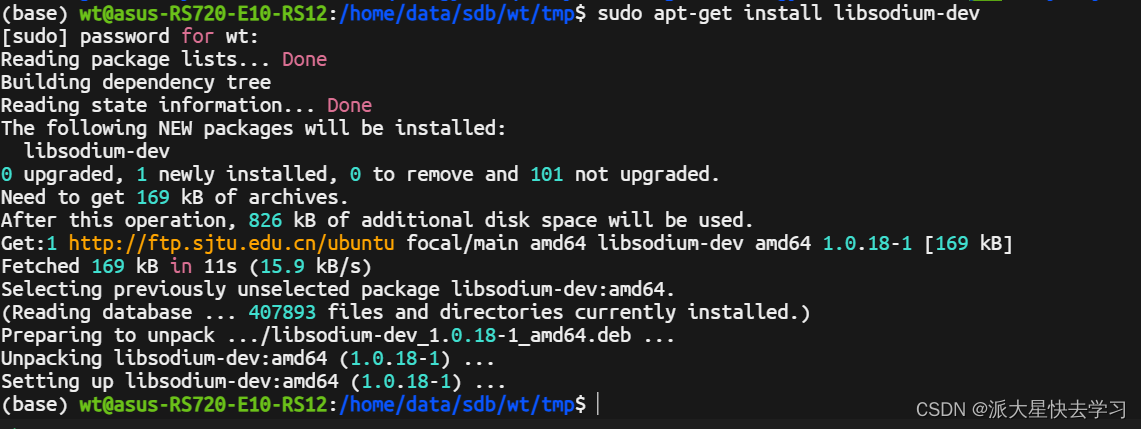
devtools::install_github("YosefLab/VISION@v2.1.0") #Please note that the version would be v2.1.0 安装成功devtools::install_github("wu-yc/scMetabolism") #安装成功!
已经全部安装好了,进入正题!
根据GitHub上的教程走代码如下:
library(scMetabolism)
library(ggplot2)
library(rsvd)载入我自己的数据根据教程跑代码
luad_data<-readRDS("/home/data/sdb/single_cell_data/processed_single_cell/GSE131907_luad.rds")
luad_data@active.assay #SCT
luad_data<-sc.metabolism.Seurat(obj = luad_data, method = "AUCell", imputation = F,
ncores = 2, metabolism.type = "KEGG") 
然后就报错了报错内容如下

然后我去看了一下sc.metabolism.Seurath函数的解释,也没看出来什么,所以去GitHub上面看了一下这个函数的源码:scMetabolism/R/compute_metabolism_Seurat.R at main · wu-yc/scMetabolism (github.com)
之后我发现,因为seurat 更新到V5之后,它的数据存放发生了改变,所以出现了错误

这个函数里面用到的还是seurat V4的示例数据,所以只需要修改一下就好了。
不了解seurat V4数据结构的可以看一下这个链接:
Seurat对象内部结构简介 - 谢大飞的文章 - 知乎 https://zhuanlan.zhihu.com/p/665483959

也就是说其实需要的数据是单细胞的稀疏矩阵,seurat V5将原始counts矩阵放在了
countexp<-obj@assays$RNA@layers$counts 但是这个是没有rownames和colnames的
需要添加行名和列名所以可以将源代码中的:
#需要修改的源代码
countexp<-obj@assays$RNA@counts
#修改之后的源代码,修改方式1
countexp<-obj@assays$RNA@layers$counts
row.names(countexp)<-row.names(obj@assays$RNA$counts)
colnames(countexp)<-colnames(obj@assays$RNA$counts)
#修改方式2
countexp<-obj@assays$RNA$counts
上面两种方式都是一样的效果。
那么重点来了,我们已经知道了要具体修改哪句源码,以及修改成什么样子,但是我们要让我们的R包也修改了呀,要怎么办呢?
修改GitHub源码的流程一般是:克隆仓库到本地,在 Rstudio 中新建项目 选择 R 包,然后选择你克隆下来的文件夹;修改你需要改的文件;最后点击右上角 Build 里面的 install
先来说第一步:克隆仓库到本地,参考链接如下
GIt——怎样克隆远程仓库到本地(敲详细)_git克隆到本地-CSDN博客
首先你打开你要克隆的远程仓库。
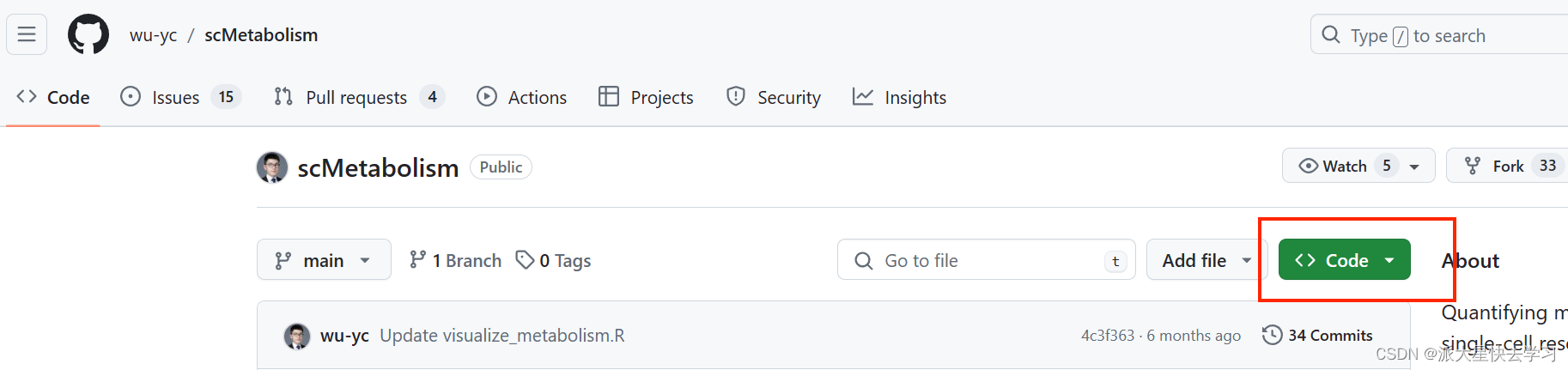

找到远程仓库的路径。在弹出的页面会发现一个Code,点击Code,会出现一个网址,复制一下这个网址,下面克隆的时候用得着。
然后打开服务器,在自己的文件夹下面创建一个文件,我创建的文件名是scMetabolism
然后再服务器linux的命令窗口输入以下命令,就是我们刚刚复制过来的网址,克隆到本地。
git clone https://github.com/wu-yc/scMetabolism.git
然后就发现刚刚创建的文件夹里面已经有了我们克隆过来的文件。
然后我们打开 Rstudio 中新建项目 选择 R 包,然后选择你克隆下来的文件夹;修改你需要改的文件;最后点击右上角 Build 里面的 install
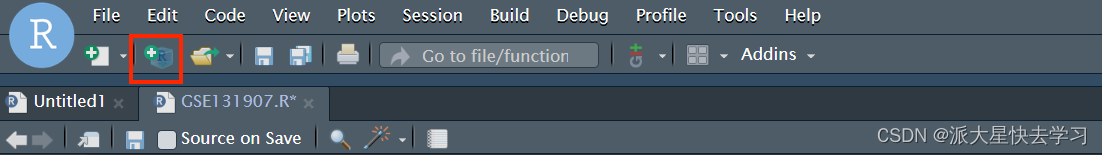
创建一个新的项目,选择existing ,也就是我们服务器上存在的文件。

然后我们就选择刚刚创建的scMetabolism文件夹就创建了项目

找到我们需要修改的函数

打开红框里面的函数这个函数就是我们要修改的,然后对要修改的代码进行修改,修改好了点击保存。
下面是我修改之后的。

然后点击Build 里面的 install--install package。这样我们就安装了修改之后的scMetabolism
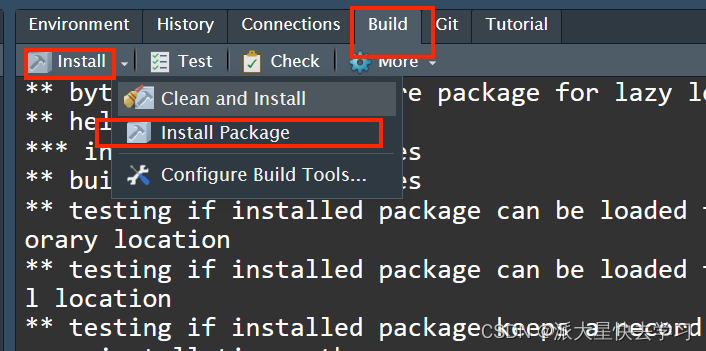
可以用命令scMetabolism::sc.metabolism.Seurat看一下是否真的修改了源码。
我这里是修改好了的。然后就可以重新跑命令啦
luad_data<-sc.metabolism.Seurat(obj = luad_data, method = "AUCell", imputation = F,
ncores = 2, metabolism.type = "KEGG")不报错啦,成功运行不过出现了warning不用管!
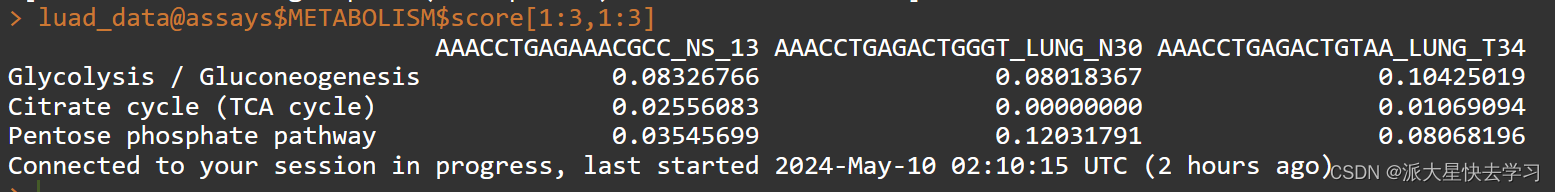
成功计算出来了每个细胞的代谢打分诶!可以进行下一步了吼吼。















 1989
1989











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









