







前言...2
实验概述...2
实验流程...2
数据下载...2
数据输入...3
质量控制...4
背景校正、标准化和汇总...6
提取表达矩阵...7
选取差异表达基因...7
聚类分析及可视化...7
差异表达基因GO注释...10
K邻近分类器...10
SVM分类...12
留一法检验-K邻近分类器...14
留一法检验-svm..15
SVM-RFE.15
SVM-RFE表达谱...17
SVM-RFE+Knn分类器...17
SVM-RFE+svm分类器...18
PCA表达谱...18
PCA+knn.19
PCA+svm..21
文章标题:The lymph node microenvironment promotes B-cell receptor signaling,NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia.
文章全文免费,可从如下网址阅读,PDF版本亦可以在程序源码文件夹下找到
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3031480/
数据组成:
由62个样本组成,皆是Affymetrix 公司的基因芯片,芯片平台[HG-U133_Plus_2]Affymetrix Human Genome U133 Plus 2.0 Array
62个白血病病人的样本中,由26个外周血peripheral blood (PB)样本,19个骨髓bone marrow (BM)样本,17个淋巴结lymph nodes (LN)样本组成。
数据下载网址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21029
研究目的:阐明白血病样本的肿瘤宿主间的相互作用造成的影响
为了阐明肿瘤宿主细胞间的相互作用造成的影响,将外周血样本分为一组,骨髓和淋巴结样本分为另外一组。
首先,下载数据至GEO数据库下载数据
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21029
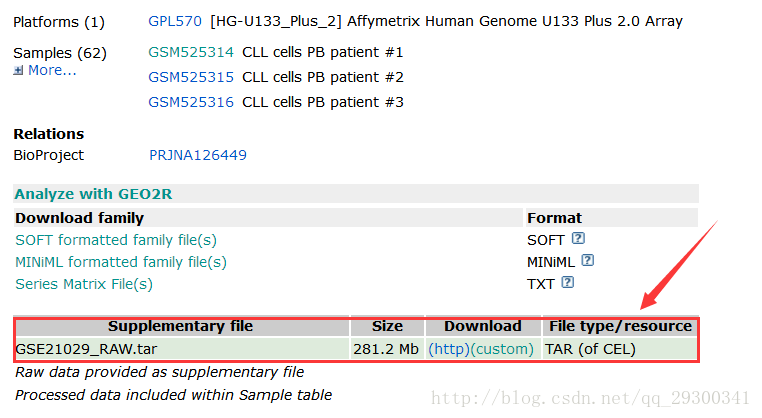
图表 1数据下载
首先,下载安装affy包,使用ReadAffy()函数读入数据,接着image()方法查看芯片的灰度图。原图PDF版可在结果文件夹“芯片灰度图.pdf”中查看。需要注意的是,Affymetrix芯片在印刷时会在四个角印制特殊的花纹,并且在左上角印制芯片的名称,花纹与芯片名称可以帮助我们借助这个图像分辨率来了解芯片数据是否可靠。
以样本 GSM525314为例画图,打开原图PDF版,可以看出来四角花纹和芯片名称非常明显,而且图像亮度适中,这给我们一个总体认识:该样本信号强度适中,比较可靠。

图表 2芯片灰度图
标准内参:mRNA是按5’端至3’端的顺序来降解的,芯片的探针组也是根据这个顺序来设计的,因此探针组的测量结果可以体现这一趋势,因为大部分的细胞都有β-actin和GAPDH基因,所以Affymetrix在大部分的芯片里都有将它们设置为一组观察RNA降解的内参基因。
而本次实验做出来的质量分析报告中显示,内参基因actin和gapdh从5’端至3’端降解的比值均在正常范围内,显示为蓝色,因此初步断定,芯片质量很好。
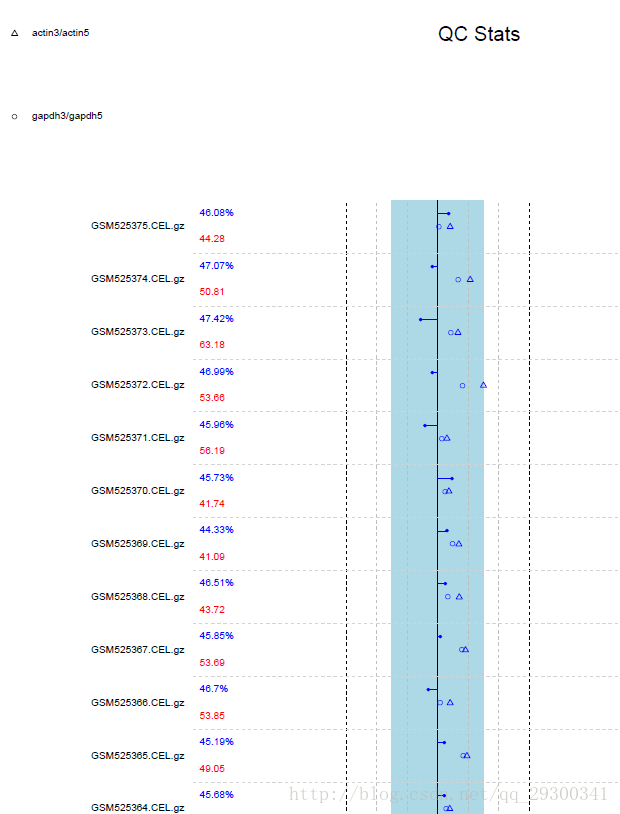
图表 3质量分析报告
在对比实验中,即使是相互比较的实验组和对照组之间,大部分基因的表达量还是应该保持一致的,平行实验之间一致性更强。相对对数表达(RLE)箱线图可以反映上述趋势,它定义为一个探针组在某个样本的表达值除以该探针组在所有样本中表达值的中位数后取对数。一个样本的所有探针组的RLE的分布可以用一个统计学中常用的箱线图表示,每个样本的中心应该非常接近纵坐标0的位置。而本次实验RLE箱线图符合此要求。
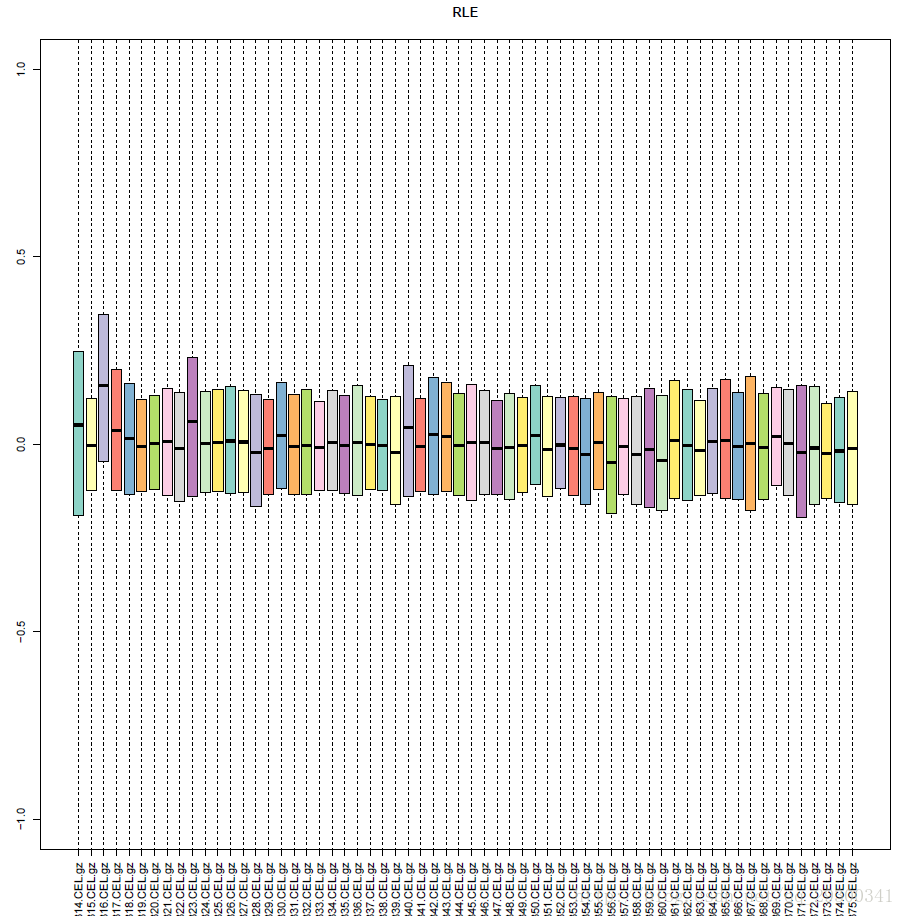
图表 4 RLE箱线图
背景校正:芯片中MM探针(序列正中央的碱基错配)的作用是检测非特异性杂交的信号。理论上,MM只有非特异性杂交,而不会有特异性杂交,MM的信号值永远小于其对应的PM(序列完全匹配)信号值。但实际中,经常发现大量的MM信号值比PM信号值还要高,因此需要应用复杂的统计模型来去除背景噪声,这个过程叫做背景校正。
标准化:标准化的目的是使各组测量或实验条件下的测量可以互相比较,消除测量间的非实验差异,非实验差异可能来源于样本制备、杂交过程或杂交信号处理等。
最后,使用一定的统计方法将前面得到的荧光强度值从探针(Probe)水平汇总到探针组(Probeset)水平,这个过程被称作汇总。
上述三步处理过程可由affy软件包中的expresso函数实现,而课上反复提及的Loess亦可作为标准化方法之一被选择。然而,实际上,使用预设参数的一体化算法更为合理高效。
下面分别使用预处理一体化算法MAS5.0、RMA、gcRMA进行预处理,并比较不同算法处理后的箱线图。可以看出来,gcRMA算法总体而言相对不错。
因此,本次实验使用的是基于RMA方法优化的gcrma方法,采用多芯片模型,(Multi-chip model)需要所有芯片一起进行标准化,背景校正的时候,基于PM的信号分布采用随机模型来估计表达值,汇总后的数据经过以2为底的对数转换。
背景校正方法:rma,标准化方法:quantile,汇总方法:medianpolish
使用exprs()函数即可提取表达矩阵,写入文件matrix.txt即可
基因表达差异的显著性分析的第一步就是选择表达具有显著性差异的基因。总体来说,这类分析的基本假设是标准化的芯片数据符合正态分布,因此所用的统计方法基本上就是T检验、F检验和方差分析及这三种统计方法的改进形式。
本次实验使用经验贝叶斯(EmpiricalBayes)方法,这种方法将标准差及信号强度的关系使用线性模型进一步强化,提高准确率,并且在计算标准差时考虑的是全部的基因,而不是排序后(人为设定的同一个窗口内)相近的基因;其次,经验贝叶斯不再局限于两组数据,可以通过设计实验对比矩阵,计算多种复杂条件下的差异表达。因此,经验贝叶斯是当前最为常用的分析方法,它已经完整地由Bioconductor中的limma包实现。
经验贝叶斯原理可见文件夹下面相应的PPT。
使用topTable函数进行筛选,为了进一步严格筛选基因,此时设定“logFC”的绝对值大于log2(1.5),即两组表达值间以2为底对数化的变化倍数要大于log2(1.5),再进行基因名注释。得到如下结果:
|
| P<=0.05; | P<=0.01; | P<=0.001; |
| 经验贝叶斯检验 | 差异表达基因贝叶斯检验_p005.txt | 差异表达基因贝叶斯检验_p001.txt | 差异表达基因贝叶斯检验_a0001.txt |
| 探针个数 | 690 | 593 | 439 |
| 基因个数 | 439 | 380 | 286 |
差异表达的基因会导致对应此基因的多个探针信号不同,因此基因个数会略小于探针个数,此表格也显示出这一性质,与事实相吻合。
备注:有些探针在其对应的GPL文件中也没有对应的基因名,因此没有注释上基因名
差异基因注释后的下一步就是统计分析和可视化。这里根据前面的分析结果,调用pheatmap包来绘制差异表达谱热图。由于就算是P<0.001且log2 Foldchange>log2(1.5),也会有286个基因,因此此处仅绘制前20个差异表达基因的热图。
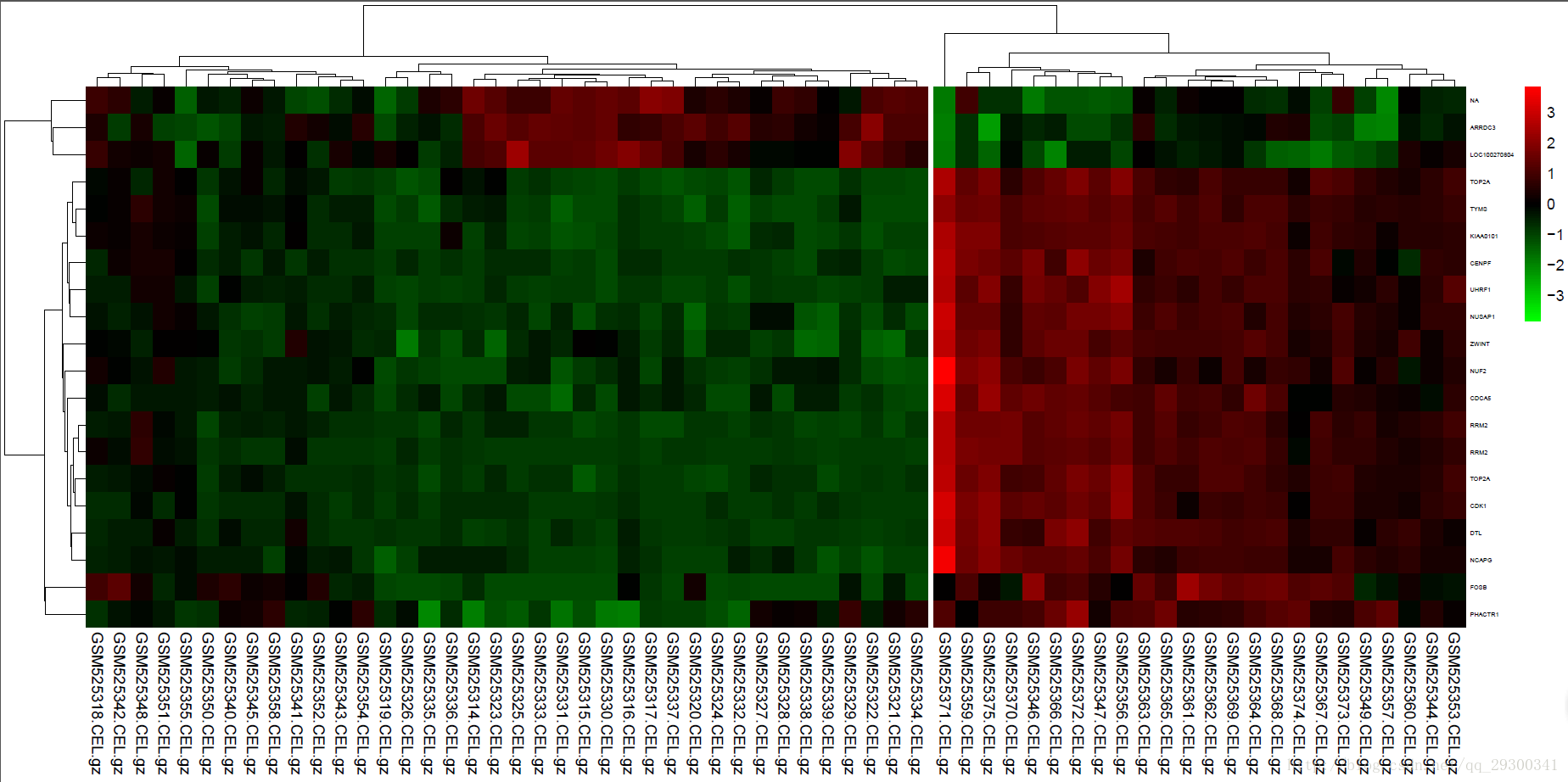
图表 5差异表达基因热图
绘制热图的同时,进行了聚类,可以看出来,聚类效果出奇的好,整体从左到右可以分成三大类,编写了一个python小脚本查看样本对应的组织类型。
19个骨髓bone marrow (BM)样本基本都在图的最左边,26个外周血peripheral blood (PB)样本基本都在图中间,17个淋巴结lymph nodes (LN)样本都在图最右边。虽然一开始是划分成两类,即PB和BM/LN对比,聚类结果也能分成三类,聚类效果很棒。
下面根据P<0.001全部差异表达基因,分别单独对样本和基因聚类。
首先单独对样本进行聚类,红框标注的是26个外周血peripheral blood (PB)样本,可以看出来基本聚类到了一起。
接着对基因聚类,由于有多个探针对应一个基因,而由图显示,这多个探针是聚类在一起,反向验证了这次实验的正确性。
对样本和基因聚类的PDF可在中间数据文件夹中找到。
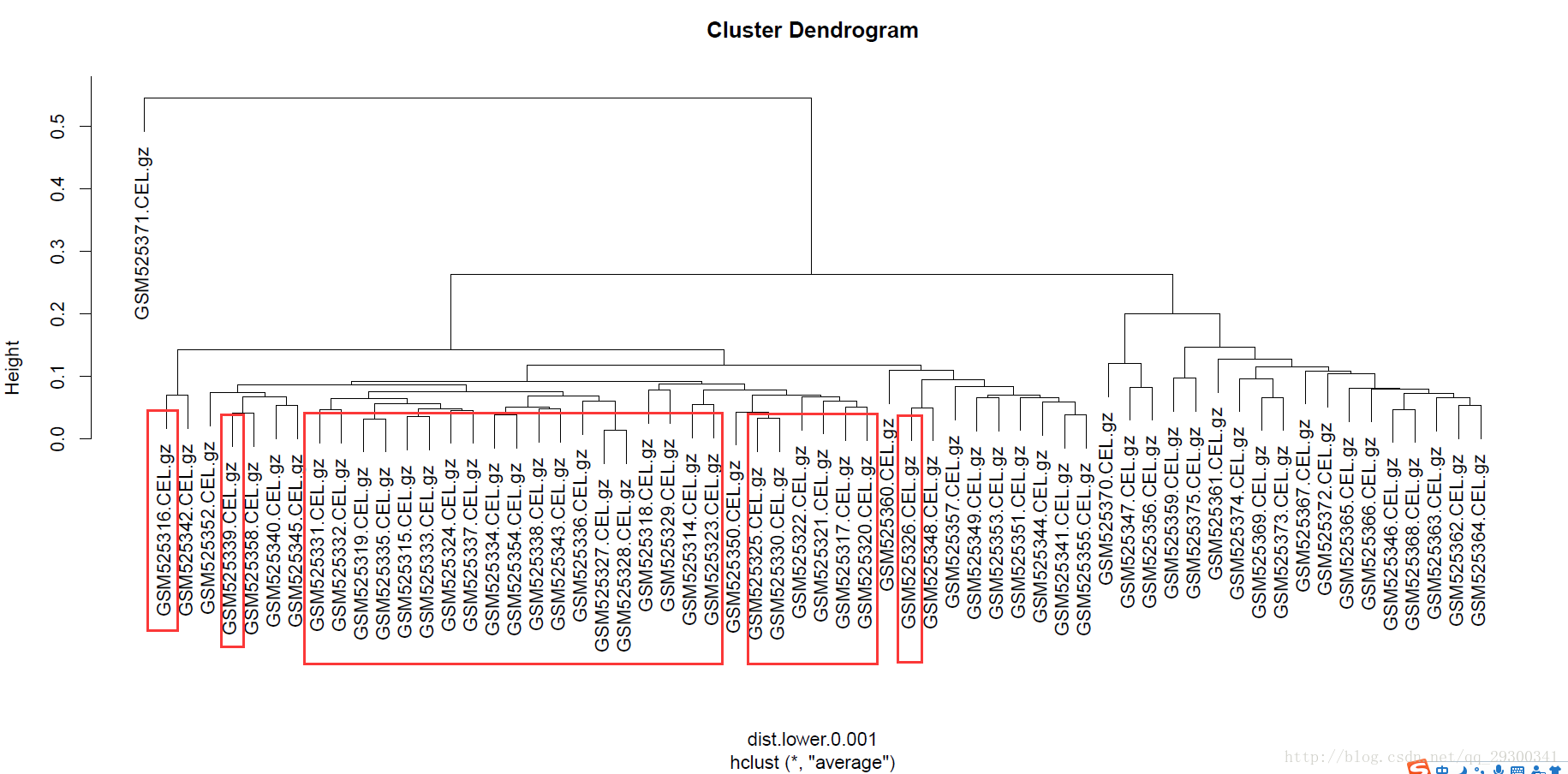
图表 6对样本聚类
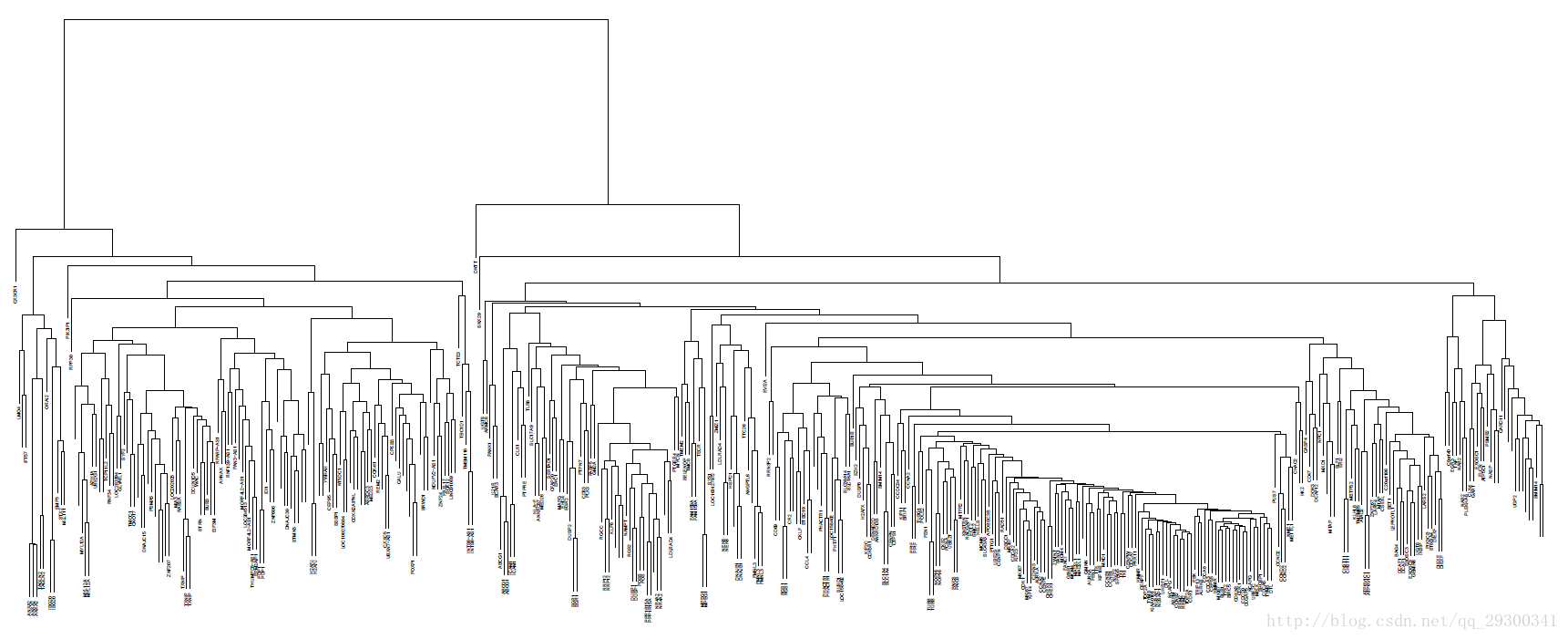
图表 7对基因聚类
提取样本P<0.001的差异表达基因的数据,根据这部分基因,绘制PCA图,可以明显看出来,两组样本具有紧致性,可以利用这些基因将两组样本分开,为后续使用“贝叶斯检验寻找差异表达基因+ K邻近法”构造分类器,提供了理论基础。
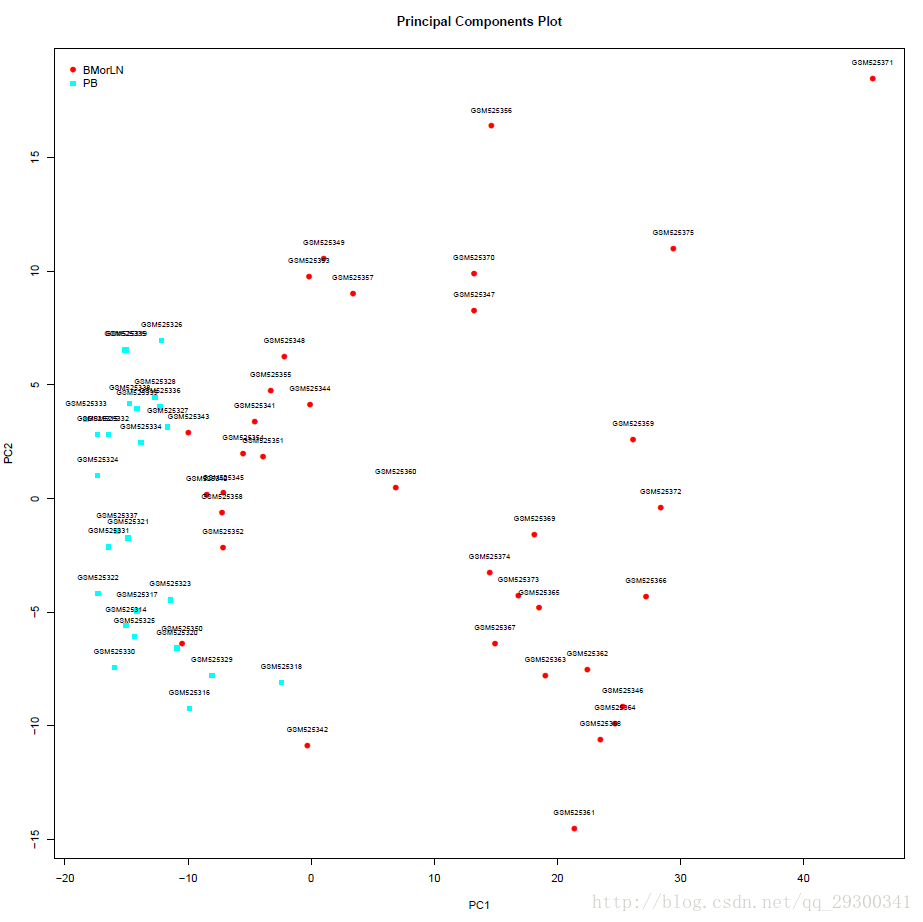
图表 8差异表达基因的样本PCA图
差异表达基因GO注释
对于P<0.001且log2 Fold change>log2(1.5)的差异表达基因,进行GO功能注释中的生物过程(BiologicalProcess)富集分析,并采用超几何检验,共有263个条目明显富集(p<0.001),明显富集的GO term详细信息可以参见结果文件夹中的GOp0.001.html
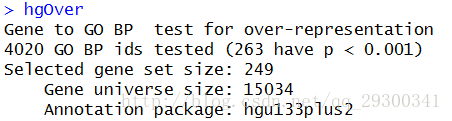
图表 9 GO注释检验
K邻近分类器
首先,使用sample函数,对表达矩阵进行3:1的取样,然后,对训练集,进行线性拟合及贝叶斯检验,寻找差异表达的基因,在P<0.001且log2 Foldchange>log2(1.5)的情况下,共筛选出281个有显著差异的探针,作为该数据的特征。
接下来,运用k近邻方法在测试集上进行分类,对分类器进行测试,依次使用k=1,2,3,4,5,6,7,8
发现当k增大到5的时候,分类效果最好。
使用CrossTable函数进行统计

图表 10二元分类器的评价
分类器相关参数评估:
| 参数 | 公式 | 值 |
| ACCuracy | (TP+TN)/(TP + TN + FP +FN) | 100% |
| Sensitivity | TP /(TP + FN) | 100% |
| Specificity | TN /(TN + FP) | 100% |
| False Positive Rate(假正率, FPR) | FP /(FP + TN) | 0% |
| False Negative Rate(假负率,FNR) | FN /(TP + FN) | 0% |
| 查准率(precision) | TP/(TP+FP) | 100% |
| 召回率(Recall) | TP/(TP+FN) | 100% |
二元分类器成功地将18个测试集样本正确分类
由于原来是三类组织的样本,进而此时也可以使用三元分类器
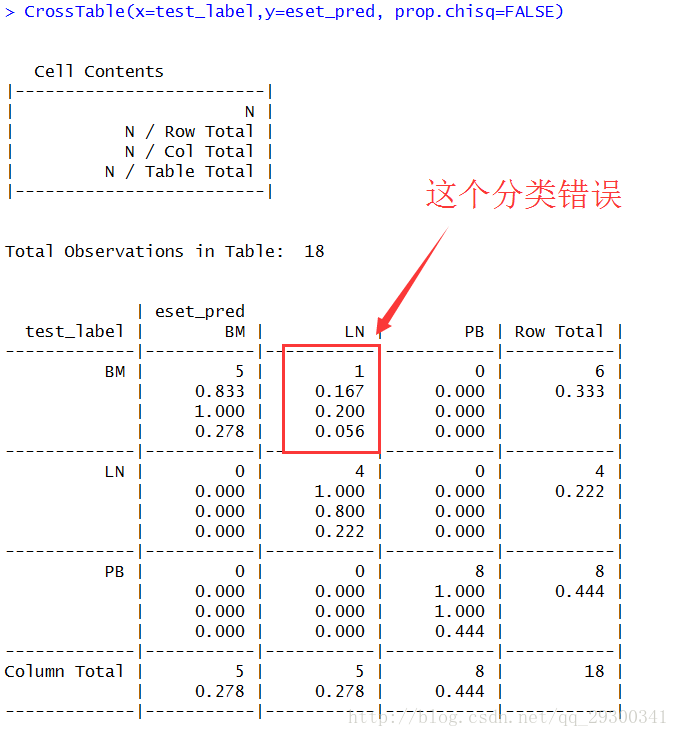
图表 11三元分类器的评价
分类器相关参数评估:(PS:三类的分类器Sensitivity和Specificity的参数需要加上一个β的协变量,不好计算)
ACCuracy =(TP+TN)/(TP + TN + FP +FN)=94.4%
准确度达到94.4%,而且,发现对PB和BM/LN进行对比,找出的差异表达的基因,居然可以很好地将PB,BM,LN三类分开,这另一方面展现出分类器良好的性能,也反映了这些差异表达基因可以作为将不同组织分开最为显著的特征,有着很棒的组织特异性,后续分析的着重点可以落在这部分基因上,阐述其作用机制。
SVM分类
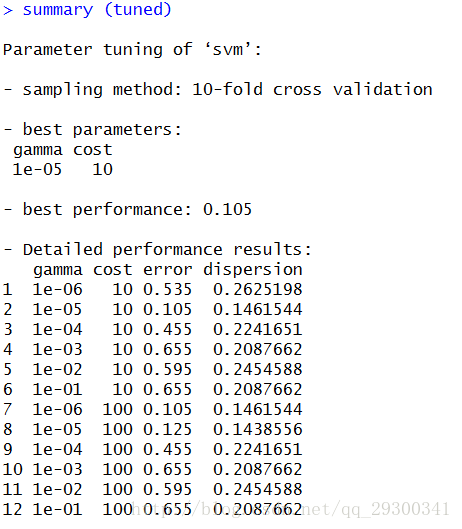
由于SVM需要较多的样本,使用sample函数取样的时候,按照4:1进行训练集和测试集的划分。对于训练集,使用tune.svm方法,取样进行交叉验证10-foldcross validation,寻找最佳的gamma和cost值,并且核函数选择径向基核函数,最小程度避免过拟合。
找出在gamma=1e-05,cost=10的情况下,参数最佳。
此时error=0.105,dispersion=0.146
PS:
1、径向基核函数:exp(-gamma*|u-v|^2),比线性核函数(u'*v)运用较广一些
2、由于此步骤为随机取样,因此不同每次得到的参数会有所不同
接下来,根据训练集,建立SVM模型。共有41个Support Vector
一共只有49个样本建立模型,就有41个作为支持向量,支持向量所占比例过大(超过80%),这表明所用特征中有不相关和冗余的特征存在,建议使用特征选择方法(RFE,文档后面也做了)进行剔除冗余和不相关者,降低维数,再用SVM。
也可以考虑重新优化参数,可是鉴于已经用tune.svm寻参,找到的参数并不好;不如使用定参,后面用RFE进行特征筛选。
计算矩阵的MDS值(Classical multidimensional scaling)进行主成分分析,使用ggplot2绘制主成分图,并将支持向量(Support Vector)标注出来。
MDS值的定义:https://en.wikipedia.org/wiki/Multidimensional_scaling
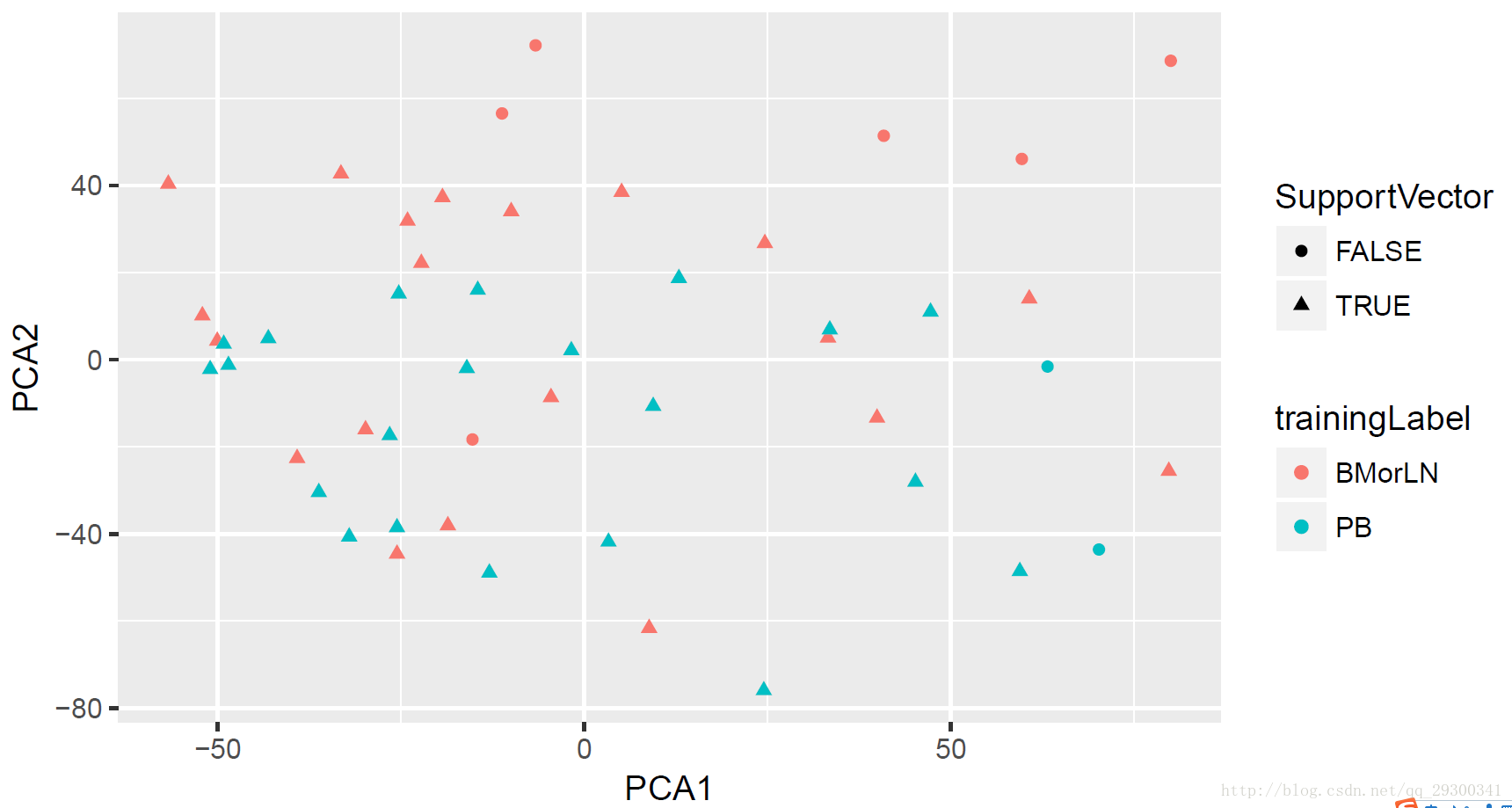
图表 13 SVM模型PCA分析
然后,使用此模型,对测试集数据进行分类,使用列联表统计正确率
| testLabel | BMorLN | PB |
| BMorLN | 8 | 2 |
| PB | 0 | 3 |
ACCuracy=84.6%
BMorLN为positive,PB为negative,有两个BMorLN被分成了PB
分类器相关参数评估:
| 参数 | 公式 | 值 |
| ACCuracy | (TP+TN)/(TP + TN + FP +FN) | 84.6% |
| Sensitivity | TP /(TP + FN) | 100% |
| Specificity | TN /(TN + FP) | 60% |
| False Positive Rate(假正率, FPR) | FP /(FP + TN) | 40% |
| False Negative Rate(假负率,FNR) | FN /(TP + FN) | 0% |
| 查准率(precision) | TP/(TP+FP) | 80% |
| 召回率(Recall) | TP/(TP+FN) | 100% |
留一法检验-K邻近分类器
首先,使用留一法,对样本进行“训练集vs测试集”的划分。每次取出一个样本作为测试集,其余样本作为训练集。然后,对训练集,调用R语言里面的limma包,进行线性拟合及贝叶斯检验,寻找差异表达的基因,在P<0.001且log2 Foldchange>log2(1.5)的情况下,筛选出有显著差异的探针,作为该数据的特征。
接下来,运用k近邻方法在测试集上进行分类,对分类器进行测试,依次使用k=1,2,3,4,5,6,7,8
发现当k在5-8的时候,分类效果都很好,最后选用k=5。
依次记录62次“训练集vs测试集”的划分后分类器预测的结果,使用CrossTable函数进行统计
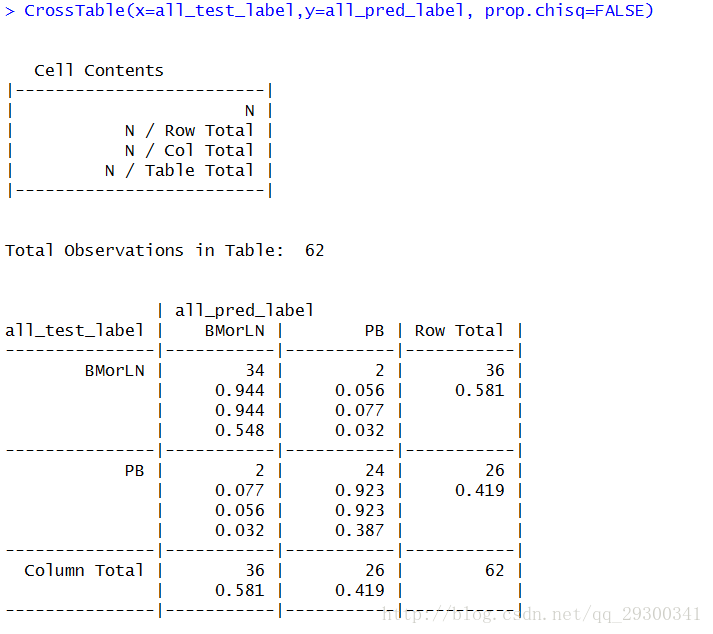
图表 14 留一法对knn评估
分类器相关参数评估:
| 参数 | 公式 | 值 |
| ACCuracy | (TP+TN)/(TP + TN + FP +FN) | 93.5% |
| Sensitivity | TP /(TP + FN) | 94.4% |
| Specificity | TN /(TN + FP) | 92.3% |
| False Positive Rate(假正率, FPR) | FP /(FP + TN) | 7.7% |
| False Negative Rate(假负率,FNR) | FN /(TP + FN) | 5.6% |
| 查准率(precision) | TP/(TP+FP) | 94.4% |
| 召回率(Recall) | TP/(TP+FN) | 94.4% |
首先,使用留一法,对样本进行“训练集vs测试集”的划分。每次取出一个样本作为测试集,其余样本作为训练集。
然后,对训练集,参考之前的参数选择:gamma=1e-05,cost=10并且核函数选择径向基核函数,构建svm分类器,在测试集的那一个样本上进行预测。
依次记录62次“训练集vs测试集”的划分后分类器预测的结果,使用CrossTable函数进行统计

图表 15 留一法对svm评估
分类器相关参数评估:
| 参数 | 公式 | 值 |
| ACCuracy | (TP+TN)/(TP + TN + FP +FN) | 91.9% |
| Sensitivity | TP /(TP + FN) | 94.4% |
| Specificity | TN /(TN + FP) | 88.5% |
| False Positive Rate(假正率, FPR) | FP /(FP + TN) | 11.5% |
| False Negative Rate(假负率,FNR) | FN /(TP + FN) | 5.6% |
| 查准率(precision) | TP/(TP+FP) | 91.9% |
| 召回率(Recall) | TP/(TP+FN) | 94.4% |
Knn和svm经过留一法检验后,可以看出来,由于本次实验样本数较少,比起之前4:1划分“训练集vs测试集”来检验svm分类器,留一法检验的时候,参与训练的样本会更多,因而构建的svm分类器也会效果更好,相关评估也会更加合理。
使用特征选择算法:SVM-RFE,进行特征选择。根据SVM在训练时生成的权向量w来构造特征排序系数,每次迭代去掉一个排序系数最小的特征,最终得到所有特征属性的递减排序。此处调用Python中的机器学习包sklearn (scikit),将表达矩阵导入,可以获得特征属性排序,即基因重要性的排序文件。
将此排序文件与之前经验贝叶斯找出来的P<0.001且log2 Fold change>log2(1.5)差异表达基因进行结合,取出SVM-RFE排名前50的基因,绘制散点图,TRUE表示这个基因在SVM-RFE排名靠前,同时也是经验贝叶斯找出来的基因。FALSE表示这个基因仅仅在SVM-RFE中排名靠前,不是经验贝叶斯找出来的差异表达基因。
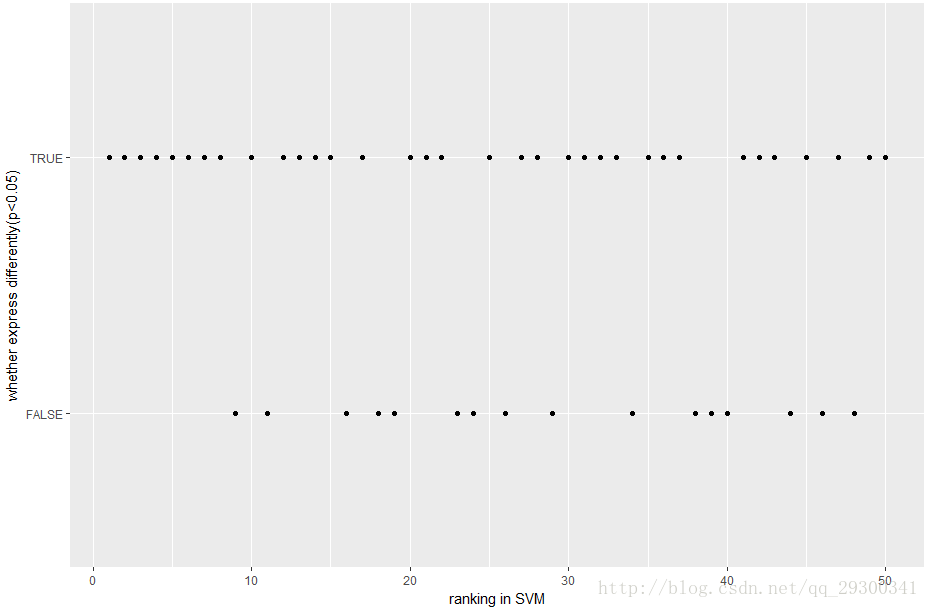
图表 16 SVM-RFE前50名的基因
接下来按照SVM-RFE找出的顺序,对全部基因是否在p<0.05的差异表达基因集中绘制散点图。
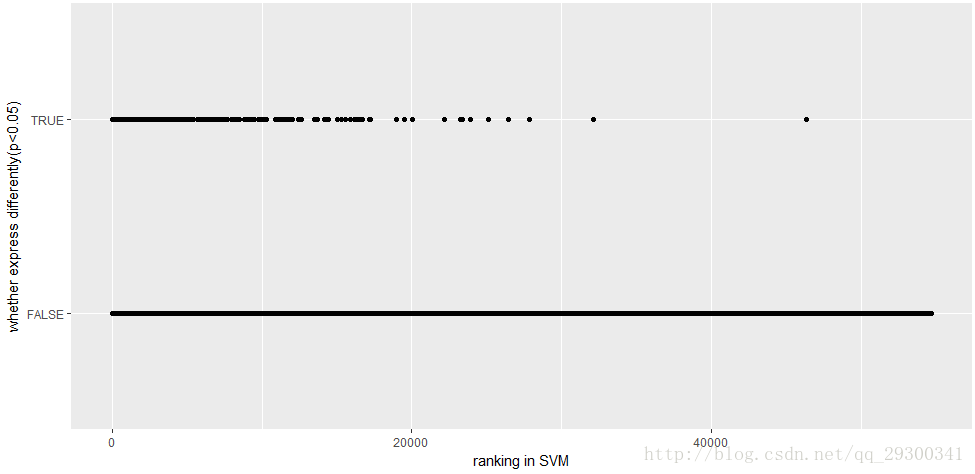
图表 17 SVM-RFE全部基因
从图中可以明显发现:
1、 从“图表16 SVM-RFE前50名的基因”可以看出来,排名前50大部分都是经验贝叶斯找出来的差异表达基因。
2、 从“图表17 SVM-RFE全部基因”可以看出来,经验贝叶斯找出来的差异表达基因排名都很高。
这两点再次验证了前面找出来的差异表达基因的正确性,同时对于两者重叠部分的基因,在统计上明显更有意义,生物学方面也会更重要,可以用于后续分析。
SVM-RFE表达谱
对SVM-RFE前50名的基因,绘制基因表达热图。
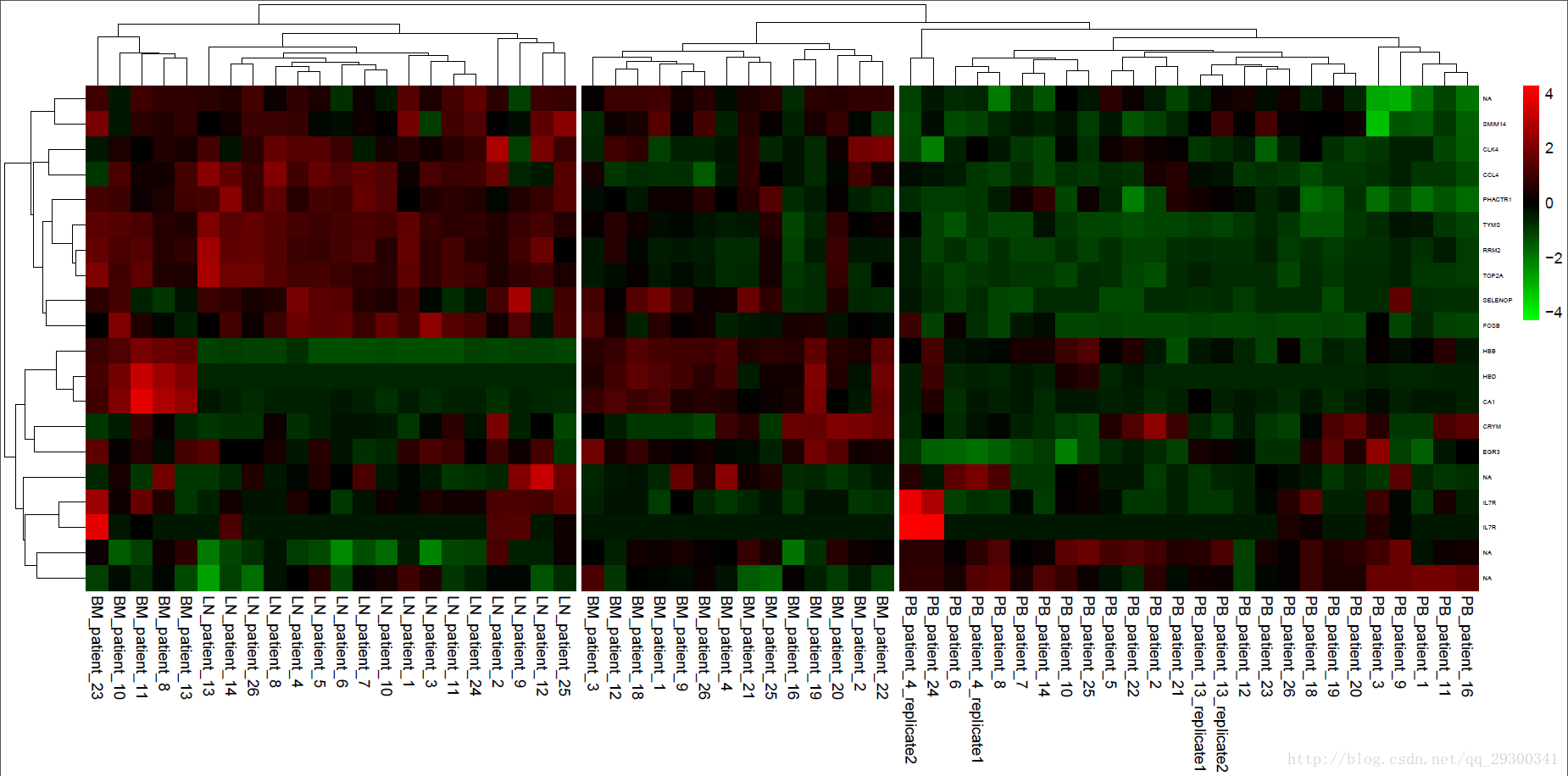
图表 18 SVM-RFE表达谱
绘制热图的同时,进行了聚类,可以看出来,聚类效果也非常好,整体从左到右可以分成三大类。
17个淋巴结lymph nodes (LN)样本大体上在图最左边,19个骨髓bone marrow (BM)样本基本都在图的最中边,26个外周血peripheral blood (PB)样本全都在图最右边。使用SVM-RFE的时候,一开始是划分成两类,即PB和BM/LN对比,聚类结果也能分成三类,聚类效果很好。
SVM-RFE+Knn分类器
选用SVM-RFE排名前50的基因,构建KNN分类器,对不同k值的分类器,计算正确率,并且绘制正确率图,正确率都非常高,比“经验贝叶斯+KNN分类器”还要略高一些。
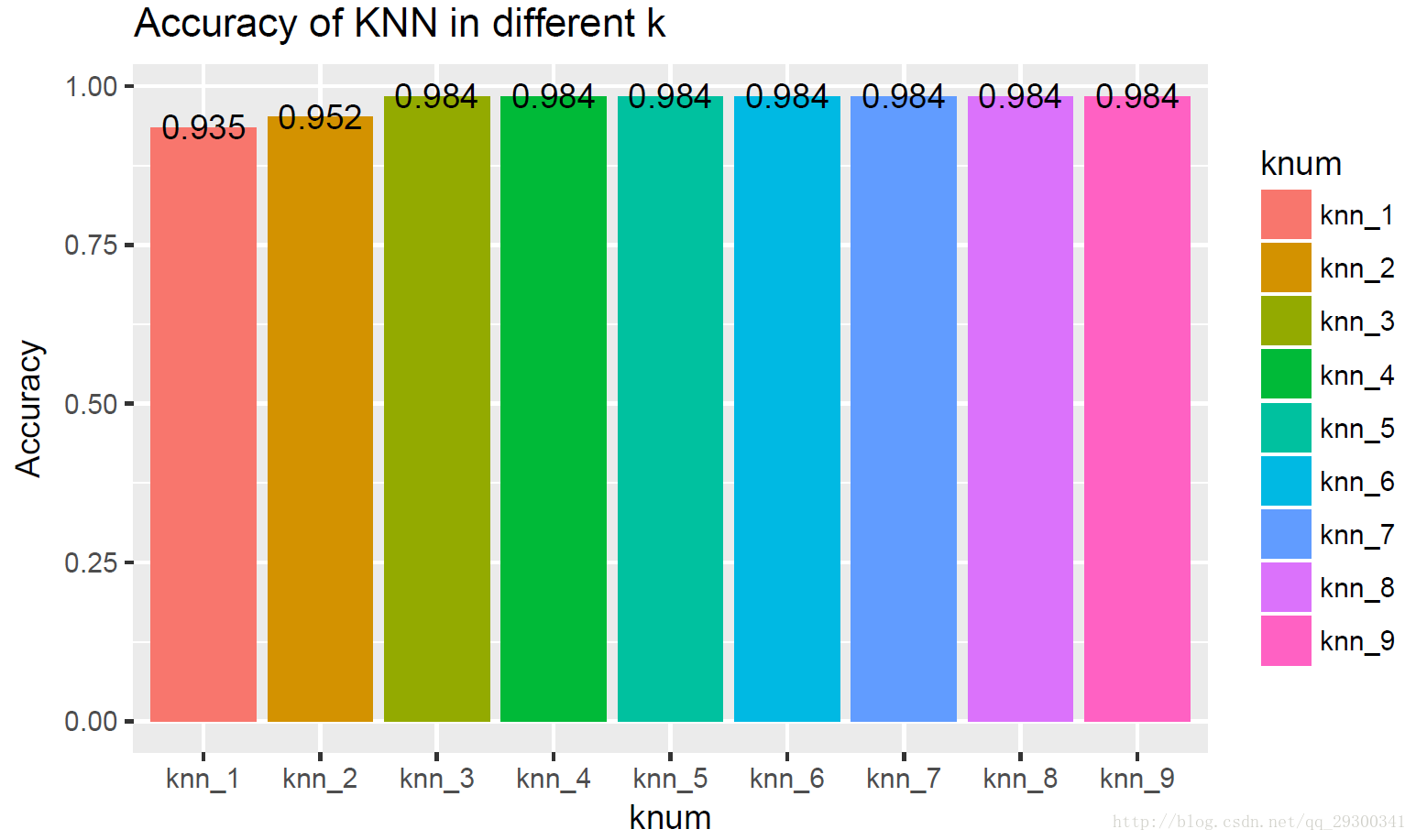
图表 19SVM-RFE+KNN准确度
SVM-RFE+svm分类器
选用SVM-RFE排名前50的基因,构建svm分类器,对svm分类器不同参数,计算正确率,并且绘制正确率图,有的参数状态下,svm正确率非常高,一度可以达到100%的正确率
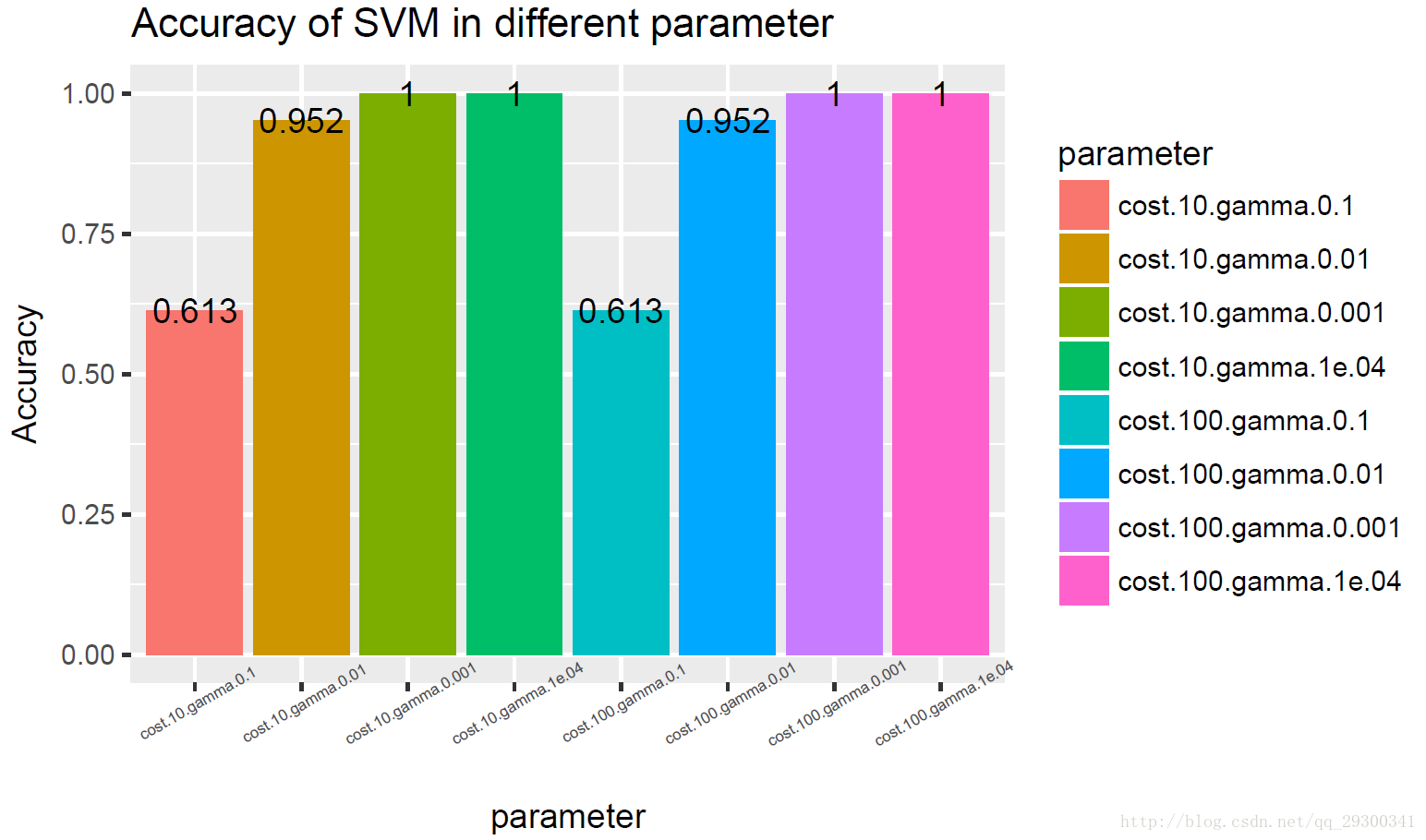
图表 20SVM-RFE+svm准确度
PCA表达谱
使用R语言里的prcomp()函数,对表达矩阵进行主成分分析,并且绘制陡坡图。
PS:陡坡图,降序显示与分量关联的特征值。用在主成分分析中,以直观地评估哪些分量占数据中变异性的大部分。 碎石图中的理想模式是一条陡曲线,接着是一段弯曲,然后是一条平坦或水平的线。建议保留陡曲线中在开始平坦线趋势的第一个点之前的那些分量。
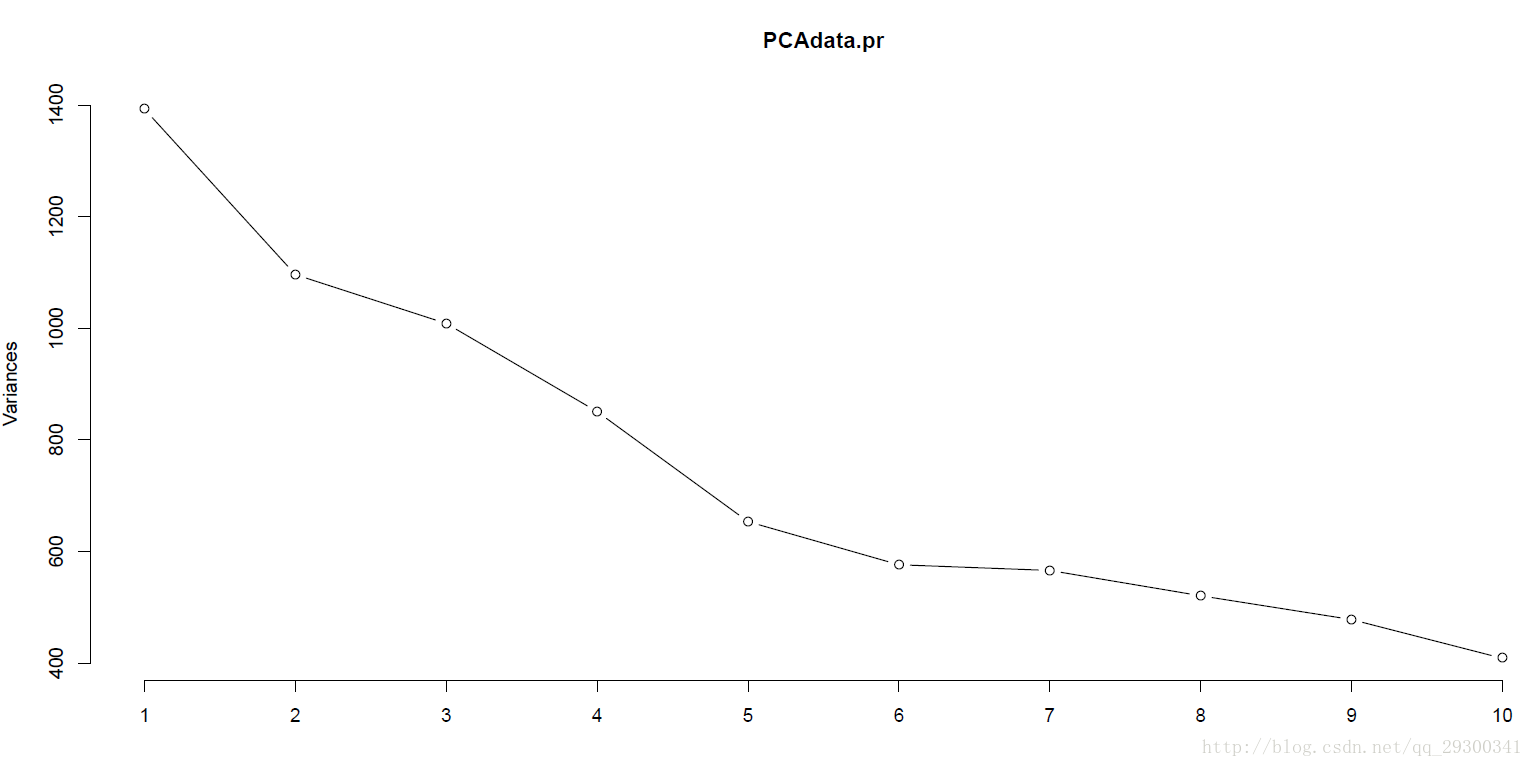
图表 21 碎石图
横坐标为主成分数量,纵坐标为方差,可以看出来大体上在第6个主成分后有平缓的趋势,表明前6个主成分就能包含大部分的信息。
下面对于前十个主成分绘制主成分表达谱。
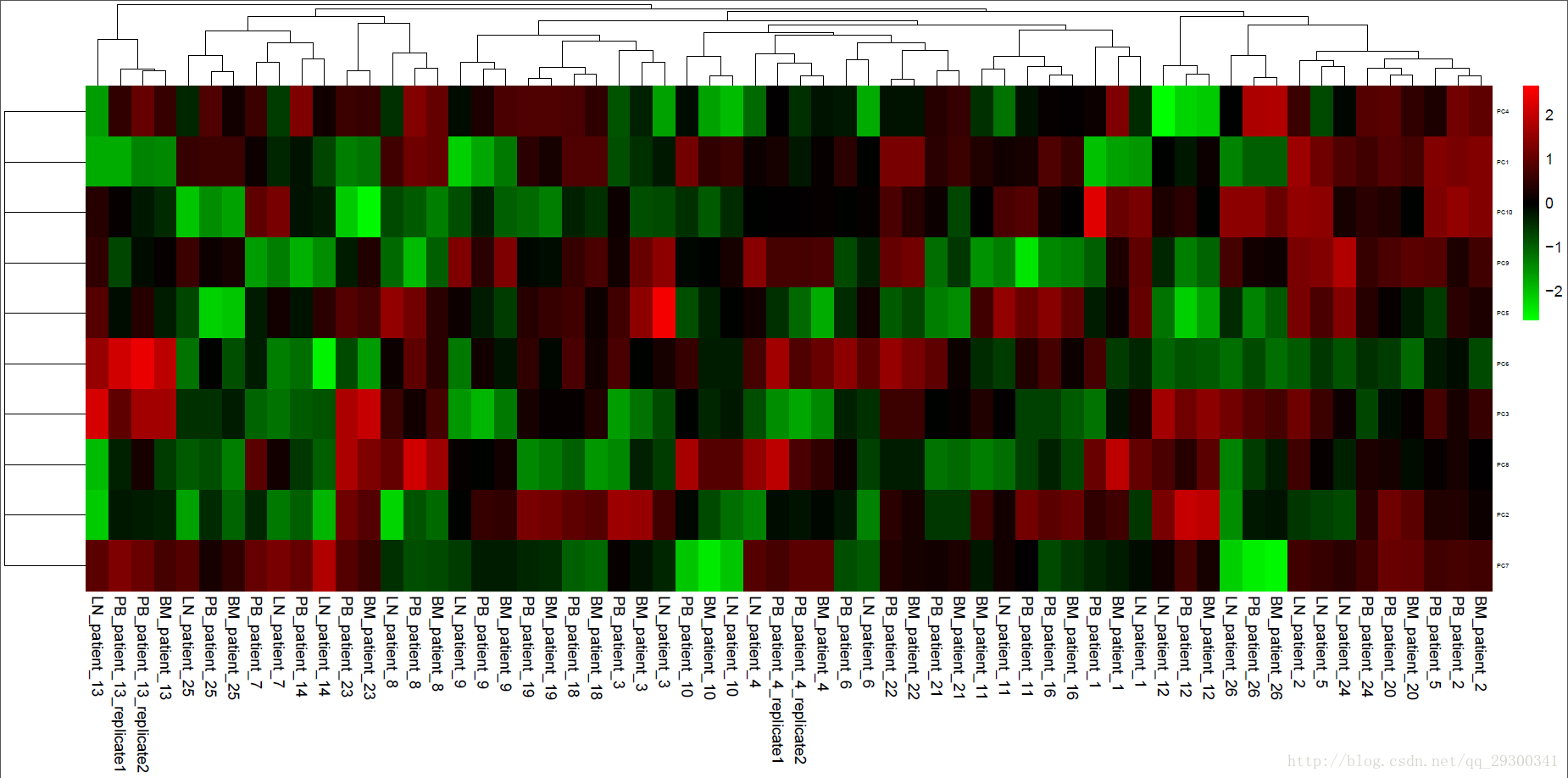
图表 22 主成分表达谱
ID 号注释:PB_patient_18代表着来源于第18个病人的外周血(PB)组织
从聚类分析的整体结果来看,LN、BM和PB三种组织的样本根本就不能很好分开。
理论上讲,如果总体上三组数据是分开的,那么说明我们关注的基因的组织特异性表达在组织间起到了主导作用,如果不是,很可能是其他其他因素起到主导作用,就比如这次实验,数据的实验样本来源于不同的病人,很可能是个体差异起到了主导性因素,而聚类结果也显示出这一特点,对所有基因进行主成分分析并且聚类,来源同一病人的样本会聚类到一起去,而不是来源于同一组织的样本会聚到一起去。
主成分分析之后,发现从来源是否是同一病人这一角度来看,主成分数据有较好的紧致性,可以用于分类,而从LN、BM和PB三种组织的角度来看,主成分数据并不具备很好的紧致性。
接下来,尝试构建KNN和svm分类器,选取前10个主成分作为数据。
首先,使用留一法,对样本进行“训练集vs测试集”的划分。每次取出一个样本作为测试集,其余样本作为训练集。接下来运用k近邻方法在测试集上进行分类,对分类器进行测试,依次使用k=1,2,3,4,5,6,7,8,9,10
每个样本被压缩为10个数据值,发现当k在5-10的时候,分类效果稍微会好一些,大致在65%左右,根据奥卡姆剃刀定律原则,模型越简单越好,最后选用k=5。
依次记录62次“训练集vs测试集”的划分后分类器预测的结果,使用CrossTable函数进行统计。

图表 23留一法对PCA+knn评估
分类器相关参数评估:
| 参数 | 公式 | 值 |
| ACCuracy | (TP+TN)/(TP + TN + FP +FN) | 59.7% |
| Sensitivity | TP /(TP + FN) | 72.2% |
| Specificity | TN /(TN + FP) | 42.3% |
| False Positive Rate(假正率, FPR) | FP /(FP + TN) | 57.7% |
| False Negative Rate(假负率,FNR) | FN /(TP + FN) | 27.8% |
| 查准率(precision) | TP/(TP+FP) | 63.4% |
| 召回率(Recall) | TP/(TP+FN) | 72.2% |
k=5,PCA选取前10个,正确率只有59.7%,比起之前“筛选差异表达基因+KNN”的效果要差了很多。
进一步探索k近邻分类器k值和主成分数量对正确率的影响,绘制k从1-10,主成分数为2-62时候Accuracy(正确率)的变化折线图。
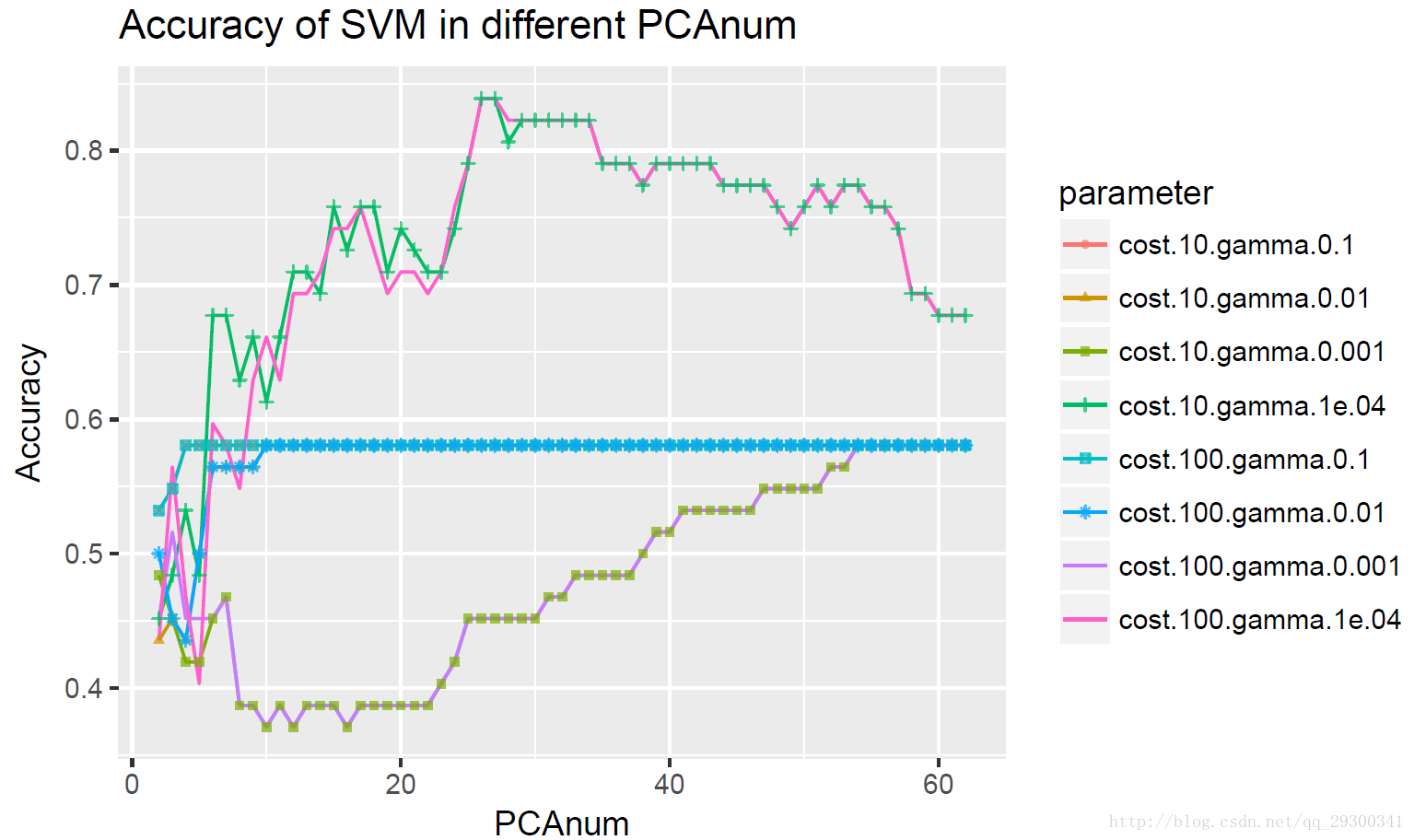
图表 24 k值和主成分数量对Accuracy的影响
很明显发现对于K近邻分类器,主成分数量越多,信息越完整,正确率也越高。
其中k>5都有很好的分类正确率,最高可以达到80%
首先,使用留一法,对样本进行“训练集vs测试集”的划分。每次取出一个样本作为测试集,其余样本作为训练集。
然后,对训练集,经过一系列尝试,选择参数:gamma=1e-04,cost=100并且核函数选择径向基核函数,构建svm分类器,在测试集的那一个样本上进行预测。
依次记录62次“训练集vs测试集”的划分后分类器预测的结果,使用CrossTable函数进行统计

图表 25留一法对PCA+svm评估
分类器相关参数评估:
| 参数 | 公式 | 值 |
| ACCuracy | (TP+TN)/(TP + TN + FP +FN) | 66.1% |
| Sensitivity | TP /(TP + FN) | 72.2% |
| Specificity | TN /(TN + FP) | 57.7% |
| False Positive Rate(假正率, FPR) | FP /(FP + TN) | 42.3% |
| False Negative Rate(假负率,FNR) | FN /(TP + FN) | 27.8% |
| 查准率(precision) | TP/(TP+FP) | 70.3% |
| 召回率(Recall) | TP/(TP+FN) | 72.2% |
PCA选取前10个正确率只有66.1%,比起之前用全部数据进行构建分类器,现在只输入10个主成分,效果要差了很多。
进一步探索主成分数量对svm分类器正确率的影响,绘制不同svm参数条件下,主成分数为2-62时候Accuracy(正确率)的变化折线图,可以明显发现主成分数量越多,信息越完整,正确率也越高。
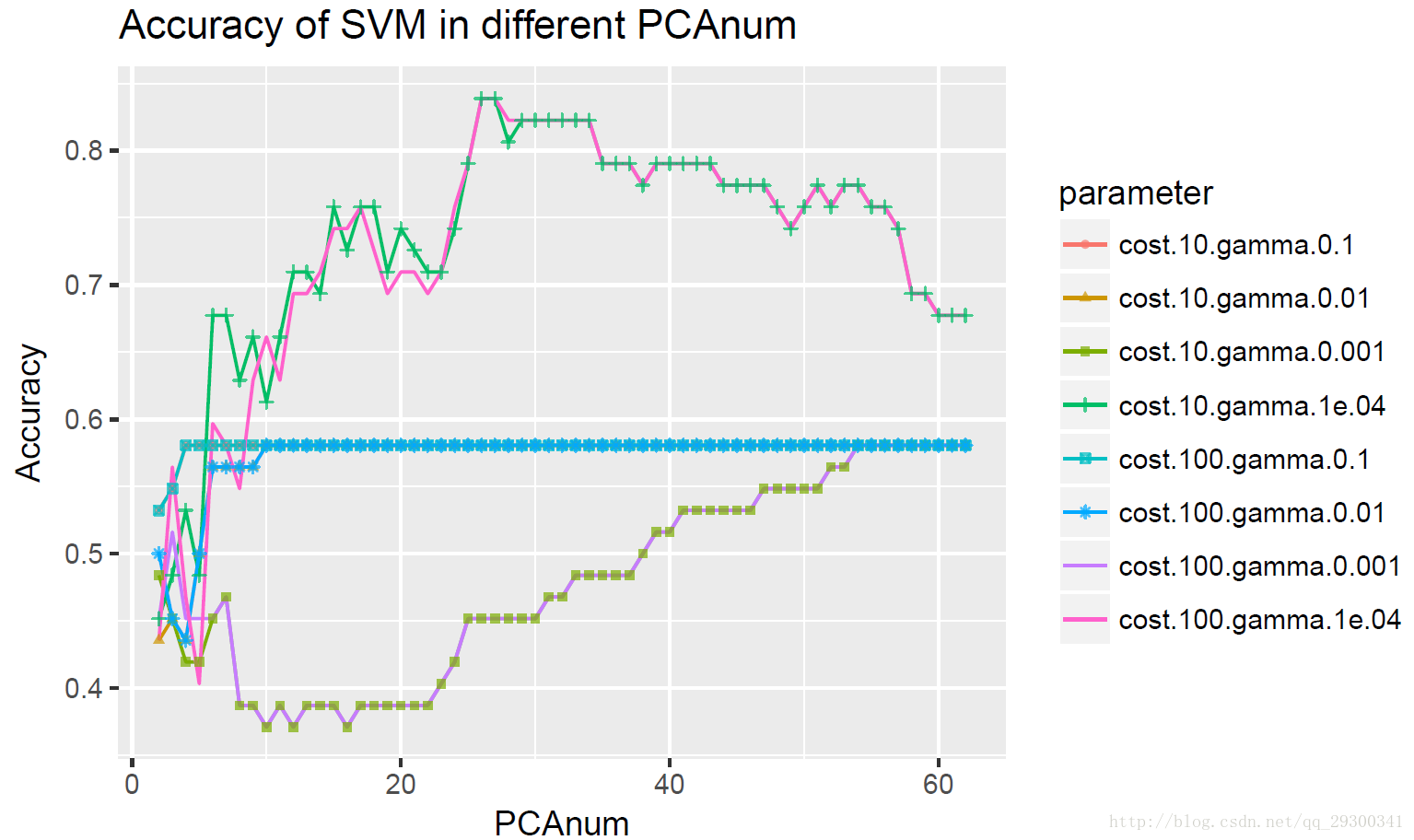
图表 26svm参数和主成分数量对Accuracy的影响
当cost=100,gamma=0.1,正确率保持在58.06%,明显是欠拟合,因为正样本占总样本的比例正好是36÷62=58.06%,分类器将所有样本都分为正类。
当主成分数量只要达到一定的数量,正确率就能达到最高的将近90%,高于KNN+PCA分类器。
综上,最好的特征选取方法是经验贝叶斯和SVM-RFE,用两者选取特征构建出的分类器,用留一法检验都能达到90%以上的正确率,使用PCA以后,缺失了太多的信息,分类器构建效果不佳;而对于分类器,svm的稳定性和参数有很大关系,好的参数会比knn效果好很多,而参数调整得不好,容易欠拟合。
文件代码都可以在百度云盘里下载到~欢迎交流参考
链接:https://pan.baidu.com/s/1qXGu0RM 密码:jg49
附录
phenotype.txt文件
SampleID Disease
GSM525314 PB
GSM525315 PB
GSM525316 PB
GSM525317 PB
GSM525318 PB
GSM525319 PB
GSM525320 PB
GSM525321 PB
GSM525322 PB
GSM525323 PB
GSM525324 PB
GSM525325 PB
GSM525326 PB
GSM525327 PB
GSM525328 PB
GSM525329 PB
GSM525330 PB
GSM525331 PB
GSM525332 PB
GSM525333 PB
GSM525334 PB
GSM525335 PB
GSM525336 PB
GSM525337 PB
GSM525338 PB
GSM525339 PB
GSM525340 BMorLN
GSM525341 BMorLN
GSM525342 BMorLN
GSM525343 BMorLN
GSM525344 BMorLN
GSM525345 BMorLN
GSM525346 BMorLN
GSM525347 BMorLN
GSM525348 BMorLN
GSM525349 BMorLN
GSM525350 BMorLN
GSM525351 BMorLN
GSM525352 BMorLN
GSM525353 BMorLN
GSM525354 BMorLN
GSM525355 BMorLN
GSM525356 BMorLN
GSM525357 BMorLN
GSM525358 BMorLN
GSM525359 BMorLN
GSM525360 BMorLN
GSM525361 BMorLN
GSM525362 BMorLN
GSM525363 BMorLN
GSM525364 BMorLN
GSM525365 BMorLN
GSM525366 BMorLN
GSM525367 BMorLN
GSM525368 BMorLN
GSM525369 BMorLN
GSM525370 BMorLN
GSM525371 BMorLN
GSM525372 BMorLN
GSM525373 BMorLN
GSM525374 BMorLN
GSM525375 BMorLN
phenotypeAnnotation.txt
GSM525314 PB_patient_1
GSM525315 PB_patient_2
GSM525316 PB_patient_3
GSM525317 PB_patient_4_replicate1
GSM525318 PB_patient_4_replicate2
GSM525319 PB_patient_5
GSM525320 PB_patient_6
GSM525321 PB_patient_7
GSM525322 PB_patient_8
GSM525323 PB_patient_9
GSM525324 PB_patient_10
GSM525325 PB_patient_11
GSM525326 PB_patient_12
GSM525327 PB_patient_13_replicate1
GSM525328 PB_patient_13_replicate2
GSM525329 PB_patient_14
GSM525330 PB_patient_16
GSM525331 PB_patient_18
GSM525332 PB_patient_19
GSM525333 PB_patient_20
GSM525334 PB_patient_21
GSM525335 PB_patient_22
GSM525336 PB_patient_23
GSM525337 PB_patient_24
GSM525338 PB_patient_25
GSM525339 PB_patient_26
GSM525340 BM_patient_1
GSM525341 BM_patient_2
GSM525342 BM_patient_3
GSM525343 BM_patient_4
GSM525344 BM_patient_8
GSM525345 BM_patient_9
GSM525346 BM_patient_10
GSM525347 BM_patient_11
GSM525348 BM_patient_12
GSM525349 BM_patient_13
GSM525350 BM_patient_16
GSM525351 BM_patient_18
GSM525352 BM_patient_19
GSM525353 BM_patient_20
GSM525354 BM_patient_21
GSM525355 BM_patient_22
GSM525356 BM_patient_23
GSM525357 BM_patient_25
GSM525358 BM_patient_26
GSM525359 LN_patient_1
GSM525360 LN_patient_2
GSM525361 LN_patient_3
GSM525362 LN_patient_4
GSM525363 LN_patient_5
GSM525364 LN_patient_6
GSM525365 LN_patient_7
GSM525366 LN_patient_8
GSM525367 LN_patient_9
GSM525368 LN_patient_10
GSM525369 LN_patient_11
GSM525370 LN_patient_12
GSM525371 LN_patient_13
GSM525372 LN_patient_14
GSM525373 LN_patient_24
GSM525374 LN_patient_25
GSM525375 LN_patient_26














 7万+
7万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









