






linux常用命令处理生物大数据
head -n 4 hsp.fa #查看文件前4行
grep -i '>bol' hsp.fa #''内为搜索条件,-i表示忽略大小写
grep -v '>' hsp.fa #-v表示反转结果,输出不包含>的行
cat 1.txt 2.txt >3.txt #合并两个文件
head -n 4 hsp.fa | grep '>' > number.txt #将前四行的序列号输出到一个文件中
wc -l hsp.fa #统计行数
grep '>' hsp.fa | wc -l
cut -f 3 gff #提取gff文件的第三列,-d默认分隔符为tab键(可以省略)
sort -k 3 gff #将gff文件按第三列进行排序,-t表示默认分隔符为tab键
sort -nrk 4 gff | cut -f 4 #将gff文件第四列按数值从大到小排列并将其提取出来 -r表示翻转(默认从小到大)
sort -k 3 gff | cut -f 3 |uniq -c #第三列去重复并统计
awk : -F指定分隔符(默认为空格键或tab键),$0 表示整行,$1表示第一列 $n表示第n列
awk '{print $1}' gff #输出gff文件的第一列
awk '$3== "gene" {print $0}' gff #输出第三列为gene的整行
awk ‘$3=="gene" && $4>=152 && $5<=180 {print $4"\t"$5}' gff # &&表示并且 \t表示tab键
& #后台符
jobs #查看当前终端进程运行的情况
kill %1 #终止指定进程
nohup #不挂断运行命令,与&配合使用,运行的程序可在终端关闭后继续进行
ps #查看任务运行情况
kill 73
find . -name *.fasta #查找当前目录下以.fasta结尾的文件
find . -size +2k #查找当前目录下大于2k的文件
md5sum -c md5.txt #将read1和read2的测序文件以及md5.txt放于同一个目录,运行该命令返回值为OK则数据完整
md5sum *gz > md5.txt #计算md5值
perl语言正则表达式
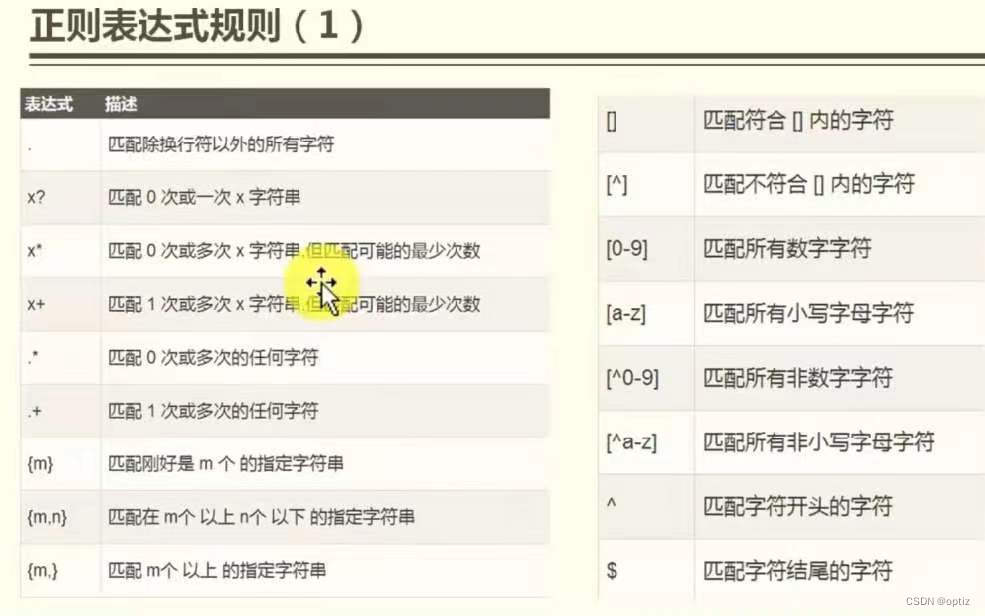
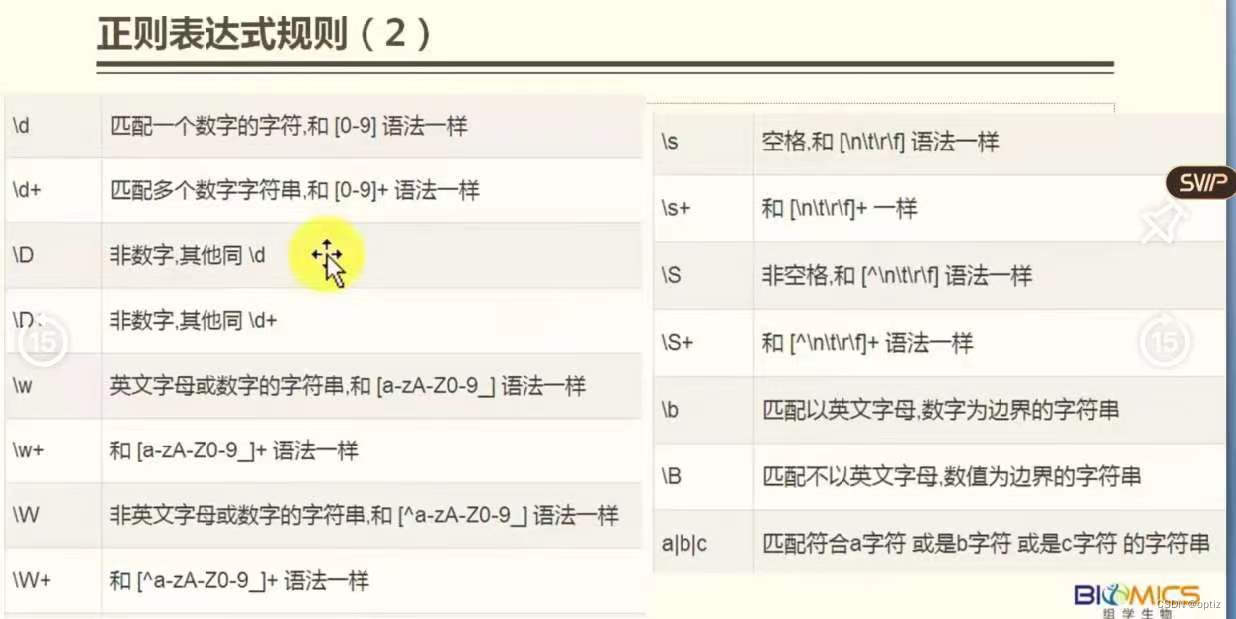

| #或
捕获Jan 1987 为Jan 1987 1987 : (/w+/s(/d+))
捕获128X456为128 456 :(/d+)X(/d+)
aspera 下载ENA数据库fastq文件
docker pull omicsclass/sratoolkit
docker run --rm -it -v /root:/work omicsclass/sratoolkit
ascp -QT -l 300m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR807/008/SRR8071498/SRR8071498_1.fastq.gz ./
批量下载
准备文件
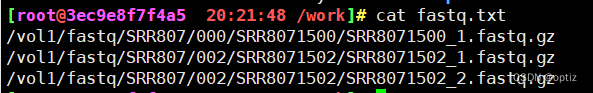
ascp -v -QT -l 300m -P33001 -k1 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh --mode recv --host fasp.sra.ebi.ac.uk --user era-fasp --file-list fastq.txt ./
# -k1表示断点续传 -i密钥 --host数据存放地址 --user表示ENA数据库的用户名
sra文件转换为fastq格式
fastq-dump --gzip --split-files SRR8071501.sra\
fasta序列处理-提取-截取序列
docker pull omicsclass/samtools
docker run --rm -it -v /root:/work omicsclass/samtools:latest
grep '>' -c 1.fa #fasta序列条数统计
fasta序列长度统计
samtools faidx 1.fa #samtools faidx 能够对fasta序列建立一个后缀为.fai的索引文件(共五列,第一列为序列名称,第二列为长度)

提取序列
samtools faidx 1.fa ERR3 >2.fa #提取1.fa文件中序列名为ERR3的序列,保存到2.fa中
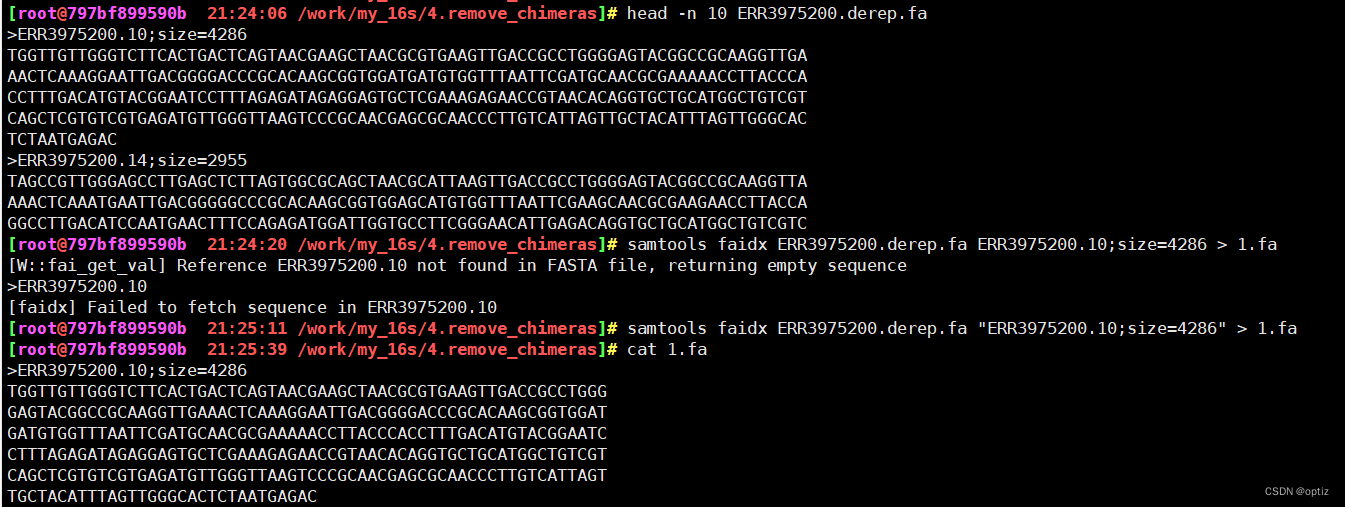
截取序列
samtools faidx 1.fa pt:2-100 #截取1.fa文件中序列号为pt的第2-100个碱基
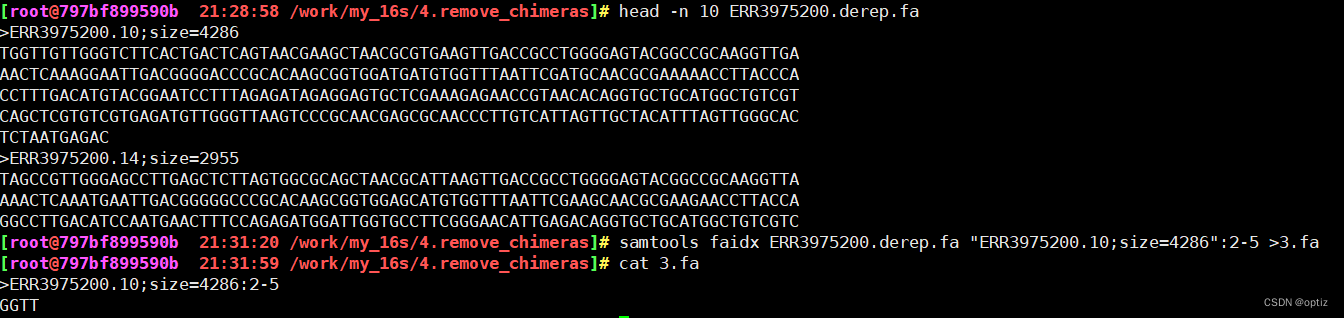
blast序列比对
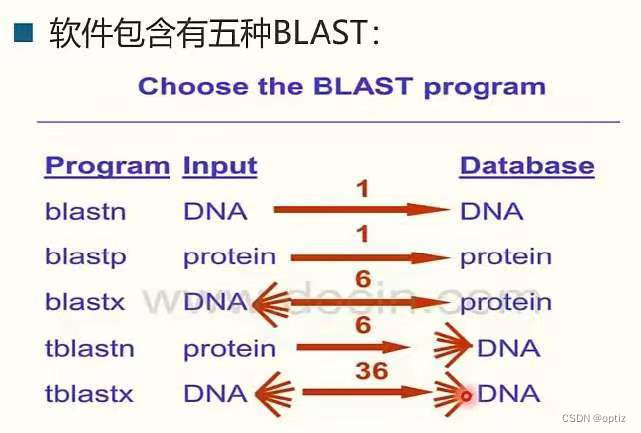
下载参考基因组
wget -c #-c表示断点续传
docker pull omicsclass/blast-plus
docker run --rm -it -v /root:/work omicsclass/blast-plus:latest
建库:每个库生成3个文件
makeblastdb -in nucl.fa -dbtype nucl -out nucl.fa #-in 输入文件 -dbtype文件类型核酸序列
makeblastdb -in nucl.fa -dbtype prot -out prot.fa #-in 输入文件 -dbtype文件类型蛋白质序列
比对(以blastp为例)
blastp -query seq.fa -out seq.blast -db prot.fa -evalue 1e-5 -outfmt 6 -num_threads 4 -max_hsps 1
#-query需要比对的文件 -out输出文件 -db建库文件 -outfmt输出文件格式 -max_hsps输出比对结果最好的序列数量(不同方法只需修改blastn)
 bit score评分越大越好
bit score评分越大越好
16S-ITS-18S
#下载多样性分析docker镜像
docker pull omicsclass/ampliseq-q1docker run --rm -it -v /root:/work -w /work omicsclass/ampliseq-q1:latest bash
#linux本机中的/root目录映射到镜像中的/work目录,-w表示指定镜像中的目录为/work,执行bash命令(执行该命令后不进入容器,目录下直接生成所得文件)
准备数据
# 准备一些路径变量,方便后续调用
####################################################
dbdir=/work/database
workdir=/work/my_16s
datadir=$workdir/data
scriptdir=$workdir/scripts
fastmap=$workdir/fastmap.txt
mkdir /work/tmp
export TMPDIR=/work/tmp #防止临时目录存储 不够export PATH=$workdir/scripts:$PATH #添加代码到环境PATH中
#各大数据库地址:
silva_16S_97_seq=$dbdir/SILVA_132_QIIME_release/rep_set/rep_set_16S_only/97/silva_132_97_16S.fna
silva_16S_97_tax=$dbdir/SILVA_132_QIIME_release/taxonomy/16S_only/97/taxonomy_7_levels.txtgreengene_16S_97_seq=$dbdir/gg_13_8_otus/rep_set/97_otus.fasta
greengene_16S_97_tax=$dbdir/gg_13_8_otus/taxonomy/97_otu_taxonomy.txtsilva_18S_97_seq=$dbdir/SILVA_132_QIIME_release/rep_set/rep_set_18S_only/97/silva_132_97_18S.fna
silva_18S_97_tax=$dbdir/SILVA_132_QIIME_release/taxonomy/18S_only/97/taxonomy_7_levels.txtunite_ITS_97_seq=$dbdir/unite_ITS8.2/sh_refs_qiime_ver8_97_04.02.2020.fasta
unite_ITS_97_tax=$dbdir/unite_ITS8.2/sh_taxonomy_qiime_ver8_97_04.02.2020.txt#选择使用的数据库
SEQ=$silva_16S_97_seq
TAX=$silva_16S_97_tax##########fastmap 准备################
cd $workdir
validate_mapping_file.py -m fastmap.txt -o validate_mapping_file_output
双端reads合并
cd $workdir #回到工作目录
mkdir 1.merge_pefor i in `cat $fastmap |grep -v '#'|cut -f 1` ;do
echo "RUN CMD: flash $datadir/${i}_1.fastq.gz $datadir/${i}_2.fastq.gz \
-m 10 -x 0.2 -p 33 -t 1 \
-o $i -d 1.merge_pe"flash $datadir/${i}_1.fastq.gz $datadir/${i}_2.fastq.gz -m 10 -x 0.2 -p 33 -t 1 -o $i -d 1.merge_pe #-m表示最小overlap,-x表示最大错误比率
done
对原始数据进行fastqc质控
cd $workdir #回到工作目录
mkdir 2.fastqc
#fastqc查看数据质量分布等
fastqc -t 2 $workdir/1.merge_pe/*extendedFrags.fastq -o $workdir/2.fastqc
#质控结果汇总
cd $workdir/2.fastqc
multiqc .
数据质控:对原始序列进行去接头,删除低质量的reads等等
cd $workdir #回到工作目录
mkdir 3.data_qc
cd 3.data_qc
#利用fastp工具去除adapter
#--qualified_quality_phred the quality value that a base is qualified.#Q
# Default 15 means phred quality >=Q15 is qualified. (int [=15])
#--unqualified_percent_limit how many percents of bases are allowed to be unqualified #比率
#--n_base_limit if one read's number of N base is >n_base_limit, 3n碱基
# then this read/pair is discarded
#--detect_adapter_for_pe 接头序列未知 可设置软件自动识别常见接头for i in `cat $fastmap |grep -v '#'|cut -f 1`; do
echo "RUN CMD: fastp --thread 1 --qualified_quality_phred 10 \
--unqualified_percent_limit 50 \
--n_base_limit 10 \
--length_required 300 \
--trim_front1 29 \
--trim_tail1 18 \
-i $workdir/1.merge_pe/${i}.extendedFrags.fastq \
-o ${i}.clean_tags.fq.gz \
--adapter_fasta $workdir/data/illumina_multiplex.fa -h ${i}.html -j ${i}.json"fastp --thread 1 --qualified_quality_phred 10 \
--unqualified_percent_limit 50 \
--n_base_limit 10 \
--length_required 300 \
--trim_front1 29 \
--trim_tail1 18 \
-i $workdir/1.merge_pe/${i}.extendedFrags.fastq \
-o ${i}.clean_tags.fq.gz \
--detect_adapter_for_pe -h ${i}.html -j ${i}.json
done#质控数据统计汇总:
python $scriptdir/qc_stat.py -d $workdir/3.data_qc/ -o $workdir/3.data_qc/ -p all_sample_qc
去除嵌合体
cd $workdir #回到工作目录
mkdir 4.remove_chimeras
cd 4.remove_chimeras#去除嵌合体(法一denovo)
for i in `cat $fastmap |grep -v '#'|cut -f 1`; do
#相同重复序列合并
vsearch --derep_fulllength $workdir/3.data_qc/${i}.clean_tags.fq.gz \
--sizeout --output ${i}.derep.fa
#去嵌合体
vsearch --uchime3_deno ${i}.derep.fa \
--sizein --sizeout \
--nonchimeras ${i}.denovo.nonchimeras.rep.fa
#相同序列还原为多个
vsearch --rereplicate ${i}.denovo.nonchimeras.rep.fa --output ${i}.denovo.nonchimeras.fadone
#根据参考序列去除嵌合体(法二:接着法一继续做)
for i in `cat $fastmap |grep -v '#'|cut -f 1`; do
vsearch --uchime_ref ${i}.denovo.nonchimeras.fa \
--db $dbdir/rdp_gold.fa \
--sizein --sizeout --fasta_width 0 \
--nonchimeras ${i}.ref.nonchimeras.fa
done















 2609
2609

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









