






一、Cibersort法(只能用于分析人种)
逆转获得原始表达值(以log2为例)
rm(list=ls(all=TRUE))
install.packages('devtools')
library(devtools)
if(!require(CIBERSORT))devtools::install_github("Moonerss/CIBERSORT")
devtools::install_github("Moonerss/CIBERSORT")
library(CIBERSORT)
library(ggplot2)#绘图
library(pheatmap)#绘制热图
library(ggpubr)#堆积比例图
library(reshape2)#数据处理
library(tidyverse)#数据处理
library(preprocessCore)
library(e1071)
library(parallel)
setwd("E:\\immunecell")
expr <- read.csv("combat_expr_control+treat.csv",row.names = 1)
if(max(expr)<50){expr<-2^expr}
数据分析
#数据检验
boxplot(expr,outline=F, notch=F , las=2)
# 均数不一致需标准化 ---------------------------------------------------------------
library(limma)
expr=normalizeBetweenArrays(expr)
boxplot(expr,outline=FALSE, notch=F , las=2)
data(LM22)#调用cibersort包中自带的LM22数据,22种免疫细胞参考marker基因表达情况
class(expr)
result <- cibersort(sig_matrix = LM22, mixture_file = expr, perm = 1000, QN = TRUE)
#expr需要为矩阵
#参数包括 sig_matrix、mixture_file、perm 和 QN。sig_matrix 参数是 CIBERSORT 软件包的内置数据,mixture_file 参数是待测样本的基因表达数据。perm 参数表示是否使用随机排列法,QN 参数是 TRUE 或 FALSE,表示是否使用质量归一化(芯片数据设置为T,测序数据就设置为F)。cibersort 函数会返回一个数据框,包含了各种免疫细胞类型的比例和基因表达数据的质量归一化结果。
result <- as.data.frame(result)#行名为样本名,列名免疫细胞名
write.table(result,"cibersort_result.txt",col.names = NA,sep = "\t")
#`P-value`越小越可信;
#Correlation原表达矩阵乘以细胞占比后的数据矩阵与原表达矩阵的相关性
#RMSE 均方根误差,越小效果越好
数据预处理(提取p<0.05的样本,删除全为0的列)最终得到矩阵data
#准备分组信息
group <- data.frame(sample=character(38),group=character(38))
group$sample <- colnames(expr)
group$group <- c(rep("control",15),rep("treat",23))
data <- cbind(result,group)
data <- data[data[,"P-value"]<0.05,]
conData <- subset(data,group=="control")
treatData <- subset(data,group=="treat")
conNum <- nrow(conData)
treatNum <- nrow(treatData)
tissue <- data$group
tissue
data <- data[,1:22]
data=as.matrix(data)PCA分析(看免疫细胞能不能区分对照组和实验组样品)
data.class=rownames(data)
data.pca=prcomp(data, scale.=TRUE)
pcaPredict=predict(data.pca)#得到pca打分
#定义数据框
PCA=data.frame(PC1=pcaPredict[,1], PC2=pcaPredict[,2], group=tissue)
PCA.mean=aggregate(PCA[,1:2], list(group=PCA$group), mean)#计算对照组和实验组的均值
#定义椭圆函数
veganCovEllipse<-function (cov, center = c(0, 0), scale = 1, npoints = 100) {
theta <- (0:npoints) * 2 * pi/npoints
Circle <- cbind(cos(theta), sin(theta))
t(center + scale * t(Circle %*% chol(cov)))
}
df_ell <- data.frame()
for(g in levels(factor(PCA$group))){
df_ell <- rbind(df_ell, cbind(as.data.frame(with(PCA[PCA$group==g,],
veganCovEllipse(cov.wt(cbind(PC1,PC2),
wt=rep(1/length(PC1),length(PC1)))$cov,
center=c(mean(PC1),mean(PC2))))),group=g))
}
#绘制PCA图形
pdf(file="PCA.pdf", width=6, height=4.5)
ggplot(data=PCA, aes(PC1, PC2)) + geom_point(aes(color=group)) +
scale_colour_manual(name="Type", values=c("blue", "red"))+
geom_path(data=df_ell, aes(x=PC1, y=PC2,colour=group), size=0.8, linetype=2)+
annotate("text", x=PCA.mean$PC1, y=PCA.mean$PC2, label=PCA.mean$group)+
theme_bw()+
theme(panel.grid.major = element_blank(), panel.grid.minor = element_blank())
dev.off()
数据可视化
result1 <- result[,1:ncol(LM22)]#将免疫细胞所在列提取出来
result1 <- result1[,apply(result1, 2, function(x){sum(x)>0})]#删除全是0的列
#在矩阵的列上应用函数:apply(X, 2, fun),其中X是矩阵,fun是要应用的函数,2表示按列应用函数。热图
法一
pdf(file="Heatmap.pdf",width = 10,height = 8)
pheatmap(result1,
color = colorRampPalette(c(rep("skyblue",3.5),"#FEFCFB",rep("#ED5467",3.5)))(50),
border="skyblue",#边框颜色
main = "Heatmap",#指定图表的标题
show_rownames = T,#是否展示行名
show_colnames = T,#是否展示列名
cexCol = 1,#指定列标签的缩放比例。
scale = 'row',#指定是否应按行方向或列方向居中和缩放,或不居中和缩放。对应的值为row, column和none。
cluster_col=T,#分别指定是否按列和行聚类。
cluster_row=F,
angle_col = "45",#指定列标签的角度。
legend = F,#指定是否显示图例。
legend_breaks=c(-3,0,3),#指定图例中显示的数据范围为-3到3。
fontsize_row = 10,#分别指定行标签和列标签的字体大小。
fontsize_col = 10)
dev.off()法二:ggplot
#数据整理
identical(rownames(result1),group$Samples)
data <- cbind(rownames(result1),result1)
colnames(data)[1] <- "Samples"
data <- melt(data,id.vars = c("Samples"))##data包含三列:sample,celltype,proportion
colnames(data) <- c('Samples','celltype','proportion')
#开始绘图
mycolors <- c('#D4E2A7','#88D7A4','#A136A1','#BAE8BC','#C757AF',
'#DF9FCE','#D5E1F1','#305691','#B6C2E7','#E8EFF7',
'#9FDFDF','#EEE0F5','#267336','#98CEDD','#CDE2EE',
'#DAD490','#372E8A','#4C862D','#81D5B0','#BAE8C9',
'#A7DCE2','#AFDE9C')
pdf(file="stacked bar chart.pdf",width = 10,height = 8)
ggplot(data,
aes(Samples,proportion,fill=celltype))+geom_bar(stat="identity",position="fill")+#x 轴是变量 Samples,y 轴是变量 proportion,条形的填充颜色由变量 celltype 决定
scale_fill_manual(values=mycolors)+#填入需要填充的颜色,22种免疫细胞
ggtitle("Proportion of immune cells")+theme_gray()+theme(axis.ticks.length=unit(3,'mm'),axis.title.x=element_text(size=11))+
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))+
guides(fill=guide_legend(title="Types of immune cells"))
dev.off() 小提琴图
library(vioplot)
outTab=data.frame()
pdf(file="vioplot.pdf", width=12, height=7.5)
par(las=1,mar=c(10,6,3,3))
x=c(1:ncol(data))
y=c(1:ncol(data))
plot(x,y,
xlim=c(0,63),ylim=c(min(data),max(data)+0.05),
main="",xlab="", ylab="Fraction",
pch=21,
col="white",
xaxt="n")
for(i in 1:ncol(data)){
if(sd(data[1:conNum,i])==0){
data[1,i]=0.00001
}
if(sd(data[(conNum+1):(conNum+treatNum),i])==0){
data[(conNum+1),i]=0.00001
}
conData=data[1:conNum,i] #??????????????????????
treatData=data[(conNum+1):(conNum+treatNum),i] #??????????????????????
vioplot(conData,at=3*(i-1),lty=1,add = T,col = 'blue') #????????????С??????(????????)
vioplot(treatData,at=3*(i-1)+1,lty=1,add = T,col = 'red') #????????????С??????(????)
wilcoxTest=wilcox.test(conData,treatData)
p=wilcoxTest$p.value
if(p<0.05){
cellPvalue=cbind(Cell=colnames(data)[i],pvalue=p)
outTab=rbind(outTab,cellPvalue)
}
mx=max(c(conData,treatData))
lines(c(x=3*(i-1)+0.2,x=3*(i-1)+0.8),c(mx,mx))
text(x=3*(i-1)+0.5, y=mx+0.02, labels=ifelse(p<0.001, paste0("p<0.001"), paste0("p=",sprintf("%.03f",p))), cex = 0.8)
}
legend("topleft",
c("Control", "Treat"),
lwd=3,bty="n",cex=1,
col=c("blue","red"))
text(seq(1,64,3),-0.05,xpd = NA,labels=colnames(data),cex = 1,srt = 45,pos=2)
dev.off()
write.table(outTab,file="Diff.cell.txt",sep="\t",row.names=F,quote=F)
提取其中一组(肿瘤/正常)绘图
data2 <- cbind(result1,group)
data2 <- data2[data2$group=='treat',]
data2 <- pivot_longer(data = data2,
cols = 1:22,
names_to = "celltype",
values_to = "proportion")
sum(data2$proportion)
pdf(file="单组箱式图.pdf",width = 10,height = 8)
ggboxplot(data = data2,
x = "celltype",#箱形图中的分组变量。
y = "proportion",
color = "black",
xlab = "Types of immune cells",#x 轴标签。
ylab = NULL,
title = "TME Cell composition",
fill = "celltype",
legend.position = "bottom",
ggtheme = theme_pubr())+ theme(axis.text.x = element_text(angle = 90,hjust = 1,vjust = 1))
dev.off() 二、基因与免疫细胞相关性热图
- 1*⭐:表示p值小于0.05,置信度为95%
- 2*⭐:表示p值小于0.01,置信度为99%
- 3*⭐:表示p值小于0.001,置信度为99.9%
- 4*⭐:表示p值小于0.0001,置信度为99.99%
- 5*⭐:表示p值小于0.00001,置信度为99.999%
library(CIBERSORT)
library(ggcorrplot)
library(tidyr)
DEG_expr <- read.csv("exp6.csv",row.names = 1)
cibersort_result <- cibersort_result[,1:22]
data <- t(DEG_expr)#行名为样本名,列名为基因名
identical(rownames(data),rownames(cibersort_result))
data <- data[,1:10]#提取想要绘图的基因名称
data <- as.data.frame(data)
corr_mat <- cor(data,cibersort_result,method = "spearson")#行名为基因名,列名为免疫细胞名
#绘图
ggcorrplot(corr_mat,
show.legend=T,#颜色图例
colors=c("#2166AC","white","#B2182B"),#相关系数矩阵颜色,白色代表0
digits=2,
lab=T)+#显示变量标签
theme(axis.text.x=element_text(angle=90,vjust=0.5,hjust=1))#修改横轴标签方向
SSGSEA法
install.packages("ggpubr")
install.packages("reshape2")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GSVA")
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("GSEABase")
install.packages("limma")
library(GSVA)
library(GSEABase)
library(limma)
library(ggpubr)
library(reshape2)
setwd("E:\\3")
mydata=read.table("limma_expr_control+treat.csv",header = T,sep = ",",check.names = F,row.names = 1)
mydata <- as.matrix(mydata)
genesetd=getGmt("immune.gmt", geneIdType=SymbolIdentifier())
ssgsea_score=gsva(mydata, genesetd, method='ssgsea', kcdf='Gaussian', abs.ranking=T)
ssgsea_result=(ssgsea_score-min(ssgsea_score))/(max(ssgsea_score)-min(ssgsea_score))
write.table(ssgsea_result,"ssgsea_result.txt",sep="\t",quote=F)热图
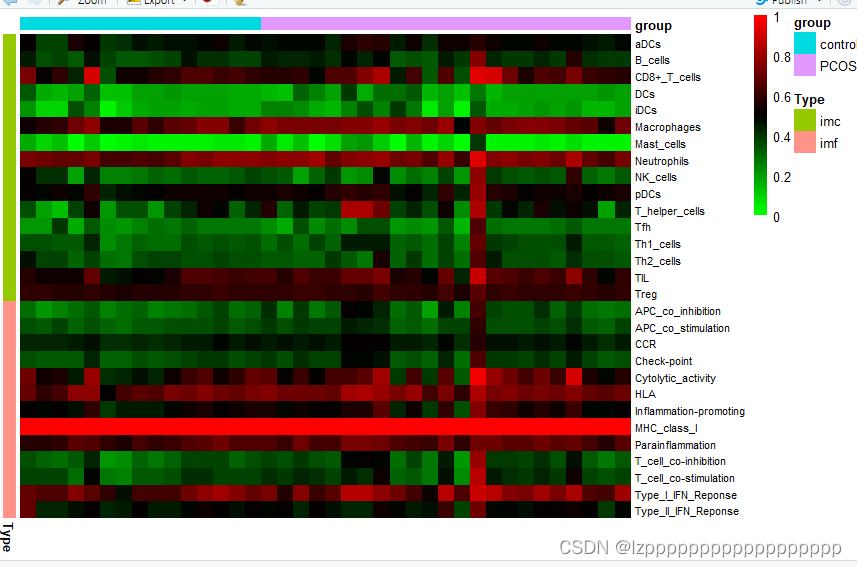
install.packages("pheatmap")
library(pheatmap)
setwd("")
data1=read.table("ssgsea_result.csv",header = T,sep = ",",check.names = F,row.names = 1)#行名免疫细胞名,列名样本名
Type<- data.frame(Type=c(rep("imc",16),rep("imf",13)))
row.names(Type) <- c("aDCs","B_cells","CD8+_T_cells","DCs","iDCs","Macrophages",
"Mast_cells","Neutrophils","NK_cells","pDCs","T_helper_cells",
"Tfh","Th1_cells","Th2_cells","TIL","Treg","APC_co_inhibition","APC_co_stimulation","CCR",
"Check-point","Cytolytic_activity","HLA","Inflammation-promoting",
"MHC_class_I","Parainflammation","T_cell_co-inhibition",
"T_cell_co-stimulation","Type_I_IFN_Reponse","Type_II_IFN_Reponse")
data2=merge(Type,data1,by=0)
data3=data2[order(data2[,"Type"],decreasing = F),]
row.names(data3) <- data3$Row.names
ht <- data3[,-c(1,2)]#行名免疫细胞名,列名样本名+Type
fenzu <- data.frame(group=c(rep("control",15),rep("PCOS",23)))
row.names(fenzu) <- colnames(ht)
pheatmap(ht,annotation_row = Type,
annotation_col = fenzu,
color = colorRampPalette(c("green", "black", "red"))(50),
cluster_cols =F,cluster_rows=F,
fontsize_row=8,
fontsize_col=6,
show_rownames = T,show_colnames = F,border_color=NA)
箱线图
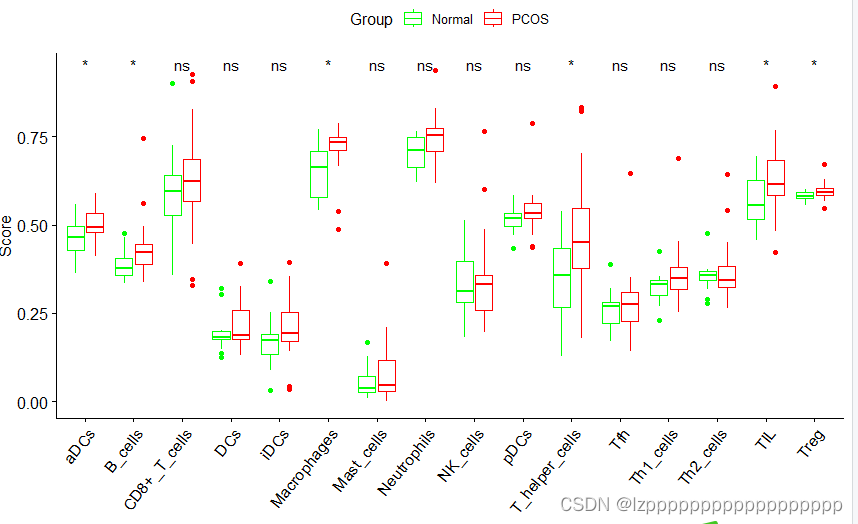

library(ggpubr)
library(reshape2)
setwd("C:\\Users\\scikuangren\\Desktop\\mynon_tumor\\imm\\step4")
data1=read.table("ssgsea_result.txt",header = T,sep = "\t",check.names =F)
data1=t(data1)
data1 <- as.data.frame(data1)
data1$mygroup <- c(rep("Normal",15),rep("PCOS",23))
imc=c("aDCs","B_cells","CD8+_T_cells","DCs","iDCs","Macrophages",
"Mast_cells","Neutrophils","NK_cells","pDCs","T_helper_cells",
"Tfh","Th1_cells","Th2_cells","TIL","Treg")
imcd=data1[,c(imc,"mygroup")]
newdata=melt(imcd,id.vars=c("mygroup"))
colnames(newdata)=c("Group","Type","Score")
a=ggboxplot(newdata, x="Type", y="Score", color = "Group",
ylab="Score",add = "none",xlab="",palette = c("green","red") )
a=a+rotate_x_text(51)
pdf("imc.pdf",8,8)
a+stat_compare_means(aes(group=newdata$Group),symnum.args=list(cutpoints = c(0,0.001,0.01,0.05, 1), symbols = c("***", "**","*", "ns")),label = "p.signif")
dev.off()
imf=c("APC_co_inhibition","APC_co_stimulation","CCR",
"Check-point","Cytolytic_activity","HLA","Inflammation-promoting",
"MHC_class_I","Parainflammation","T_cell_co-inhibition",
"T_cell_co-stimulation","Type_I_IFN_Reponse","Type_II_IFN_Reponse")
imfd=data1[,c(imf,"mygroup")]
newdata=melt(imfd,id.vars=c("mygroup"))
colnames(newdata)=c("Group","Type","Score")
a=ggboxplot(newdata, x="Type", y="Score", color = "Group",
ylab="Score",add = "none",xlab="",palette = c("blue","red") )
a=a+rotate_x_text(51)
pdf("imf.pdf",8,8)
a+stat_compare_means(aes(group=newdata$Group),symnum.args=list(cutpoints = c(0,0.001,0.01,0.05, 1), symbols = c("***", "**","*", "ns")),label = "p.signif")
dev.off()
基因与免疫细胞和免疫功能相关性热图
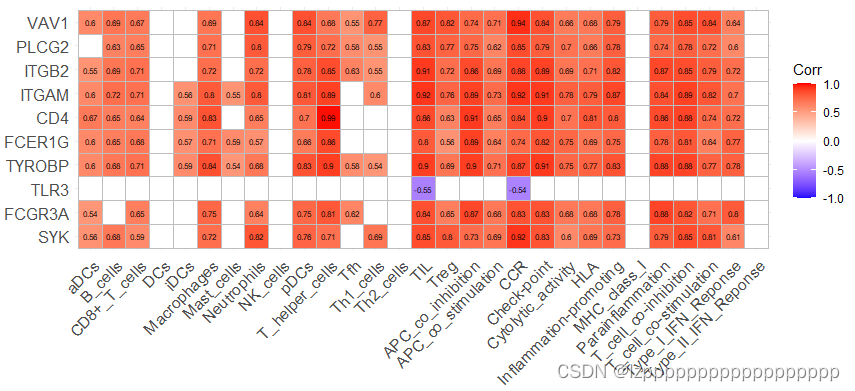
install.packages("psych")
install.packages("ggcorrplot")
library(psych)
library(ggcorrplot)
setwd("C:\\Users\\scikuangren\\Desktop\\mynon_tumor\\imm\\step5")
data1=read.table("ssgsea_result.csv",header = T,sep = ",",check.names =F,row.names = 1)
data1=t(data1) #行名样本名,列名免疫细胞名
class(data1)
data2=read.table("limma_expr_control+treat.csv",header = T,sep = ",",check.names =F,row.names=1)
mygene=c("SYK","FCGR3A","TLR3", "TYROBP", "FCER1G","CD4","ITGAM","ITGB2","PLCG2","VAV1")
data3=data2[mygene,]
data3=as.matrix(data3)
data3=t(data3) #行名样本名,列名基因名
class(data3)
mycor<- corr.test(data3,data1,method = 'spearman')
rvalue <- mycor$r
pvalue <- mycor$p
ggcorrplot(t(rvalue), show.legend = T,
p.mat = t(mycor$p.adj), digits = 2, sig.level = 0.05,insig = 'blank',lab = T, lab_size = 2)
ggcorrplot(t(rvalue), show.legend = T,
p.mat = t(mycor$p.adj), digits = 2, sig.level = 0.05,lab = T)
将相关性较高的放在上面
library(ggplot2)
library(dplyr)data <- data1
immune <- data3
outTab = data.frame()for (cell in colnames(immune)) {
if (sd(immune[,cell]) == 0) { next }
for (gene in colnames(data)) {
x = as.numeric(immune[,cell])
y = as.numeric(data[,gene])
corT = cor.test(x, y, method = "spearman")
cor = corT$estimate
pvalue = corT$p.value
text = ifelse(pvalue < 0.001, "***", ifelse(pvalue < 0.01, "**", ifelse(pvalue < 0.05, "*", "")))
outTab = rbind(outTab, cbind(Gene = gene, Immune = cell, cor, text, pvalue))
}
}outTab$cor = as.numeric(outTab$cor)
# 对Gene和Immune进行排序,使相关性最高的排在最上方和最左边
outTab <- outTab %>%
mutate(Gene = reorder(Gene, cor, FUN = max),
Immune = reorder(Immune, cor, FUN = max))ggplot(outTab, aes(Immune, Gene)) +
geom_tile(aes(fill = cor), colour = "grey", size = 1) +
scale_fill_gradient2(low = "#5C5DAF", mid = "white", high = "#EA2E2D") +
geom_text(aes(label = text), col = "black", size = 3) +
theme_minimal() +
theme(axis.title.x = element_blank(), axis.ticks.x = element_blank(), axis.title.y = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1, size = 8, face = "bold"),
axis.text.y = element_text(size = 8, face = "bold")) +
labs(fill = paste0("*** p<0.001", "\n", "** p<0.01", "\n", " * p<0.05", "\n", "\n", "Correlation")) +
scale_x_discrete(position = "bottom")
write.csv(outTab,"cor-p.csv")
ROC曲线、矫正曲线
install.packages("rms")
install.packages("ROCR")
library(rms)
library(ROCR)
setwd("C:\\Users\\scikuangren\\Desktop\\mynon_tumor\\imm\\step6")
data1=read.table("limma_expr_control+treat.csv",sep=",",header = T,row.names = 1)
mygene=c("SYK","FCGR3A","TLR3", "TYROBP", "FCER1G","CD4","ITGAM","ITGB2","PLCG2","VAV1")
data2=data1[mygene,]
data3 <-as.data.frame( t(data2))
data3$status <- c(rep(0,15),rep(1,23))
data1 <- data3
for (i in colnames(data1[,2:ncol(data1)])){
exp=factor(ifelse(data1[,i]<median(data1[,i]),"0","1"))
data1[,i]=exp
data1[,i]=factor(data1[,i],labels=c("Low","High"))
}
ddist <- datadist(data1)
options(datadist="ddist")
mylog<- glm(status~.,family=binomial(link = "logit"),data = data1)
summary(mylog)
coefficients(mylog)
exp(coefficients(mylog))#OR
#nom
mylog<-lrm(status~.,data=data1,x=T,y=T)
mynom<- nomogram(mylog, fun=plogis,fun.at=c(0,0.1,0.2,0.3,0.4,0.5,0.6,0.7,0.8,0.9,1),lp=F, funlabel="Risk of PCOS")
pdf("Nom.pdf",10,8)
plot(mynom)
dev.off()
#C-index
Cindex <- rcorrcens(data1$status~predict(mylog))
Cindex
#Calibration
mycal<-calibrate(mylog,method="boot",B=1000)
pdf("Calibration.pdf")
plot(mycal,xlab="Nomogram-predicted probability of pAF ",ylab="Actual diagnosed pAF (proportion)",sub=F)
dev.off()
#AUC
pre_rate<-predict(mylog)
ROC1<- prediction(pre_rate,data1$status)
ROC2<- performance(ROC1,"tpr","fpr")
AUC <- performance(ROC1,"auc")
print(AUC,max = 100000)
AUC<-0.846 #鏀逛负鑷繁鐨凙UC
pdf("ROC.pdf")
plot(ROC2,col="blue", xlab="False positive rate",ylab="True positive rate",lty=1,lwd=3,main=paste("AUC=",AUC))
abline(0,1,lty=2,lwd=3)
dev.off()















 6895
6895











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









