






一 定义、图形

- 基本原理:基因之间相关系数的计算→关系矩阵的建立→建立邻接矩阵,利用幂指数进行加权→无尺度网络的构建→基因模块的确定
- 注意事项:1.芯片数据或RNA-seq数据,如果是转录组数据,最好是RPKM,TPM或者其他归一化后的表达量 2.15个样品以上 3.不能多于5000个基因
- 分析目的:寻找具有协调表达(共表达)的基因组成的网络模块,探索这些基因网络模块与研究的表型/性状(如癌症与正常)之间的联系,寻找与外部信息相关的hub基因,为下一步的研究/实验设计提供指导。
- WGCNA是一种构建基因共表达网络的常用系统生物学算法,与传统的基因共表达网络算法的主要差异在WGCNA采用了软阈值β对表达矩阵进行加权。
- 分析流程:构建基因-基因相似性网络→对协同表达的基因聚类→探索外部模块与基因表达的关联→鉴定模块中的hub基因
- 构建加权共表达网络可以选择一步法(one step)或分步法(step by step)进行。 一般情况下优先选择采用简便的一步法,当想要调整得到的基因模块数目时,采用分步法就会更灵活。
目录

聚类与模块图:

模块聚类图:

拓扑矩阵热图:两侧的聚类结果是相同的,对角线的颜色越深说明联系越高,分析结果较好

二 基因表达数据、临床信息准备
- 准备文件limma_expr_control+treat.csv和fenzu.csv
- 筛选做分析的基因(方差前25%)、检查缺失值、剔除离群样本、准备临床数据、样本聚类
setwd("C:\\Users\\lexb4\\Desktop\\WGCNA\\13.GEOwgcna")
#if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
BiocManager::install(c("GO.db", "preprocessCore", "impute","limma"))
#install.packages(c("matrixStats", "Hmisc", "foreach", "doParallel", "fastcluster", "dynamicTreeCut", "survival"))
#install.packages("WGCNA")
library("WGCNA")
library("limma")
data <- read.table("limma_expr_control+treat.csv",sep = ",",header = T,row.names = 1)
datExpr0 <- t(data)
dim(datExpr0)
#筛选方差前25%的基因
m.vars=apply(datExpr0,2,var)
datExpr0 <- datExpr0[,which(m.vars > quantile(m.vars, probs = seq(0,1,0.25))[4])]
class(datExpr0)
write.csv(datExpr0,"datExpr0.csv")
datExpr0 <-read.csv("datExpr0.csv",row.names = 1)
以上部分为数据的初步处理,最终得到datExpr0(行名为样本名,列名为基因名)
###检查缺失值
gsg = goodSamplesGenes(datExpr0, verbose = 3)
gsg$allOK
if (!gsg$allOK)
{
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")))
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")))
# Remove the offending genes and samples from the data:
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
#样品聚类,删除离群样本
sampleTree = hclust(dist(datExpr0), method = "average")
pdf(file = "1_sample_cluster.pdf", width = 12, height = 9)
par(cex = 0.6)
par(mar = c(0,4,2,0))
plot(sampleTree,
main = "Sample clustering to detect outliers",
sub="",
xlab="",
cex.lab = 1.5,
cex.axis = 1.5,
cex.main = 2)
#剪切线
abline(h = 150, col = "red")#划定需要剪切的枝长
dev.off()
###删除剪切线以下的样品
clust = cutreeStatic(sampleTree, cutHeight = 150, minSize = 10)
table(clust)
keepSamples = (clust==1) #保留非离群(clust==1)的样本
datExpr0 = datExpr0[keepSamples, ] #去除离群值后的数据
#准备临床数据
traitData <- read.csv("fenzu.csv",row.names = 1)
fpkmSamples = rownames(datExpr0)#以去除离群样本的样本名
traitSamples =rownames(traitData)#全部样本名
sameSample=intersect(fpkmSamples,traitSamples)
datExpr=datExpr0[sameSample,]#行名为去除离群样本后的样本名,列名为筛选后的基因名
datTraits=traitData[sameSample,]#临床信息,行名样本名,列名Normal和Tumor
rm(traitData,fpkmSamples,traitSamples,sameSample)
#样品聚类树(图片结果解释了临床数据和基因表达量的关联程度)
sampleTree2 = hclust(dist(datExpr), method = "average")
traitColors = numbers2colors(datTraits, signed = FALSE)#用颜色代表关联度,颜色越深,代表这个表型数据与这个样本的基因表达量关系越密切。
pdf(file="2_sample_heatmap.pdf",width=12,height=12)
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
dev.off()
三 构建表达网络
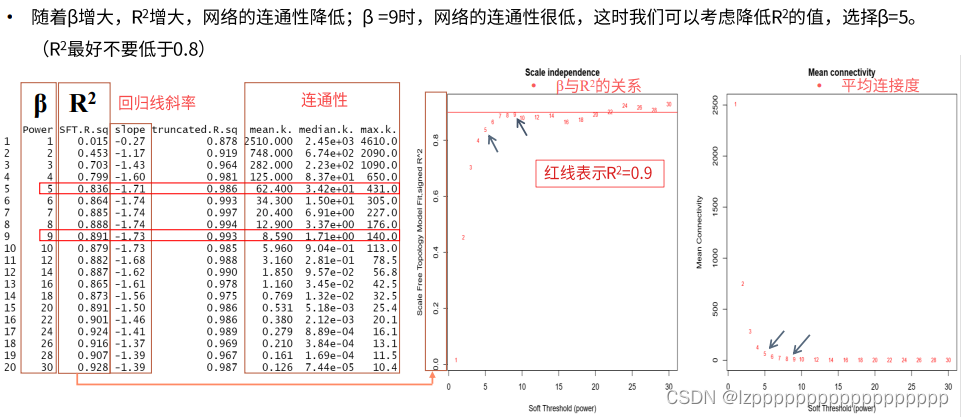
3.1 选择软阈值
options(stringsAsFactors = FALSE);
enableWGCNAThreads() #开启多线程
#计算软阈值
powers1=c(c(1:10), seq(from = 12, to=20, by=2))
RpowerTable=pickSoftThreshold(datExpr, powerVector=powers1)[[2]]
cex1=0.9
par(mfrow=c(1,2))
pdf("beta.pdf")
#beta与R2关系图(无尺度拓扑你和指数图)
plot(RpowerTable[,1],
-sign(RpowerTable[,3])*RpowerTable[,2],
xlab="Soft Threshold (power)",
ylab="Scale Free Topology Model Fit,signed R^2",
main = paste("Scale independence"),
type="n")
text(RpowerTable[,1], -sign(RpowerTable[,3])*RpowerTable[,2],
labels=powers1,cex=cex1,col="red")
abline(h=0.9,col="red")#查看位于0.9以上的点,可以改变高度值,但不能小于0.8
#连通性图
plot(RpowerTable[,1], RpowerTable[,5],
xlab="Soft Threshold (power)",
ylab="Mean Connectivity",
type="n",
main = paste("Mean connectivity"))
text(RpowerTable[,1], RpowerTable[,5], labels=powers1, cex=cex1,col="red")
dev.off()
#运行下面的代码,如果有合适的软阈值,系统会自动推荐给你。
sft <- pickSoftThreshold(datExpr, powerVector=powers1)
sft$powerEstimate
rm(sft,RpowerTable,cex1,powers1)3.2 一步法构建网络和模块检测
- 处理最大基因数为位5000,如果大于5000,这个函数会将数据集拆分为几块,这会破坏下面的一些绘图代码,即执行代码会导致错误。希望分析更大数据集的读者需要执行以下操作之一:4GB运行内存可以处理8000~10000个,16GB最多可处理20000个,32GB最多可以处理30000个。如果要分析较大的数据集,需要逐块分析。
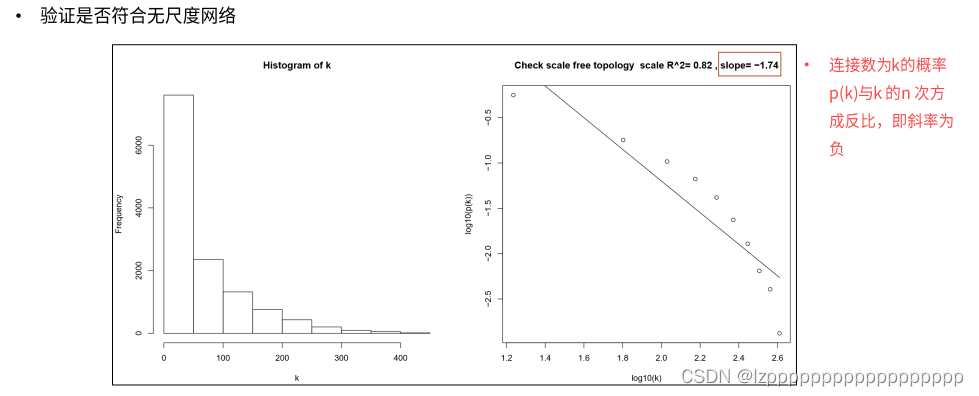
net = blockwiseModules(datExpr, power = 7,# power = 7是刚才选择的软阈值
TOMType = "unsigned", minModuleSize = 30,#minModuleSize:模块中最少的基因数
reassignThreshold = 0, mergeCutHeight = 0.25,#mergeCutHeight :模块合并阈值,阈值越大,模块越少(重要)
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,#saveTOMs = TRUE,saveTOMFileBase = "TOM"保存TOM矩阵,名字为"TOM"
saveTOMFileBase = "TOM",
verbose = 3)
#net$colors 包含模块分配,net$MEs 包含模块的模块特征基因
table(net$colors)#查看划分的模块数和每个模块里面包含的基因个数
#> 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
#> 99 609 460 409 316 312 221 211 157 123 106 100 94 91 77 76 58 47 34
#以上结果表示一共可以分为18个模块,第二行是每个模块对应的基因数,有多到少。
#从模块1开始,基因数逐渐减少。模块0是无法识别的基因数。
#模块标识的层次聚类树状图
pdf("Cluster_Dendrogram.pdf",width=12,height=9)
mergedColors = labels2colors(net$colors)
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
dev.off()
#提取分配模块和模块包含的基因信息。
moduleLabels = net$colors#各基因对应的模块(以数字形式表示)
moduleColors = labels2colors(net$colors)#各基因对应的模块颜色
MEs = net$MEs;
geneTree = net$dendrograms[[1]]
3.3 模块与表型数据关联并识别重要基因
nGenes = ncol(datExpr);
nSamples = nrow(datExpr);
# 重新计算带有颜色标签的模块
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
moduleTraitCor = cor(MEs, datTraits, use = "p");
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples);
# 通过相关值对每个关联进行颜色编码
pdf("Module-trait_relationship.pdf",width=8,height=6)
# 展示模块与表型数据的相关系数和 P值
textMatrix = paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "");
dim(textMatrix) = dim(moduleTraitCor)
par(mar = c(6, 8.5, 3, 3));
# 用热图的形式展示相关系数
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = blueWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
dev.off()
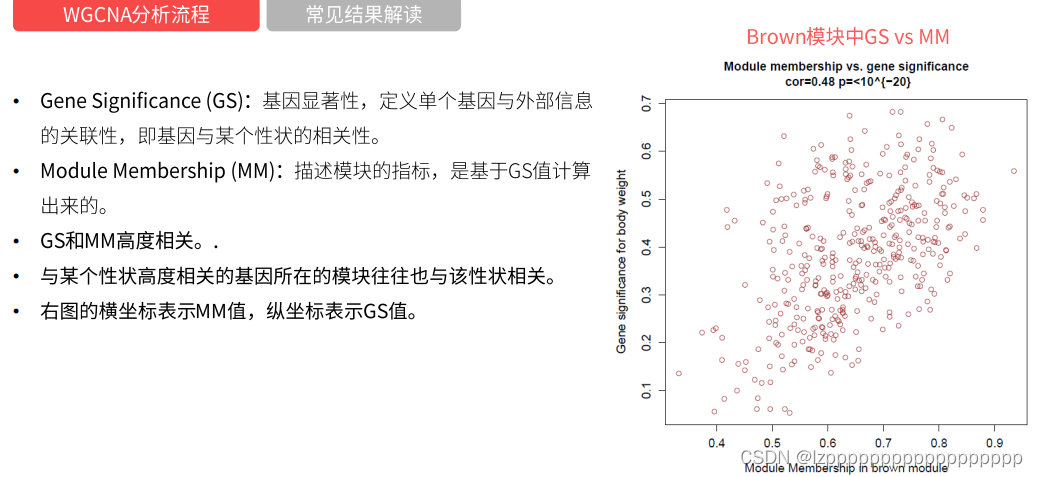
#计算MM和GS值
modNames = substring(names(MEs), 3)
geneModuleMembership = as.data.frame(cor(datExpr, MEs, use = "p"))
MMPvalue = as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples))
names(geneModuleMembership) = paste("MM", modNames, sep="")
names(MMPvalue) = paste("p.MM", modNames, sep="")
traitNames=names(datTraits)
geneTraitSignificance = as.data.frame(cor(datExpr, datTraits, use = "p"))
GSPvalue = as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples))
names(geneTraitSignificance) = paste("GS.", traitNames, sep="")
names(GSPvalue) = paste("p.GS.", traitNames, sep="")
#批量输出性状和模块散点图
for(trait in traitNames){
traitColumn=match(trait,traitNames)
for (module in modNames){
column = match(module, modNames)
moduleGenes = moduleColors==module
if (nrow(geneModuleMembership[moduleGenes,]) > 1){
outPdf=paste("9_", trait, "_", module,".pdf",sep="")
pdf(file=outPdf,width=7,height=7)
par(mfrow = c(1,1))
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, traitColumn]),
xlab = paste("Module Membership in", module, "module"),
ylab = paste("Gene significance for ",trait),
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)
abline(v=0.8,h=0.5,col="red")
dev.off()
}
}
}
#输出GS_MM数据
probes= colnames(datExpr)
geneInfo0 = data.frame(probes= probes,
moduleColor = moduleColors)
for (Tra in 1:ncol(geneTraitSignificance))
{
oldNames = names(geneInfo0)
geneInfo0 = data.frame(geneInfo0, geneTraitSignificance[,Tra],
GSPvalue[, Tra])
names(geneInfo0) = c(oldNames,names(geneTraitSignificance)[Tra],
names(GSPvalue)[Tra])
}
for (mod in 1:ncol(geneModuleMembership))
{
oldNames = names(geneInfo0)
geneInfo0 = data.frame(geneInfo0, geneModuleMembership[,mod],
MMPvalue[, mod])
names(geneInfo0) = c(oldNames,names(geneModuleMembership)[mod],
names(MMPvalue)[mod])
}
geneOrder =order(geneInfo0$moduleColor)
geneInfo = geneInfo0[geneOrder, ]
write.table(geneInfo, file = "GS_MM.xls",sep="\t",row.names=F)
#输出每个模块的基因
for(mod in 1:nrow(table(moduleColors)))
{
modules = names(table(moduleColors))[mod]
probes = colnames(datExpr)
inModule = (moduleColors == modules)
modGenes = probes[inModule]
write.table(modGenes, file =paste0("GEO_",modules,".txt"),sep="\t",row.names=F,col.names=F,quote=F)
}
利用David网站进行GO分析,得到如下表格,命名为go_vis.txt

install.packages("ggplot2")
setwd("C:\\Users\\scikuangren\\Desktop\\GO") #??Ϊ?Լ??Ĺ???Ŀ¼
inputfile="go_vis.txt"
library(ggplot2)
go=read.table(inputfile,header=T,sep="\t")
pdf("go-1.pdf")
ggplot(data=go)+geom_bar(aes(x=Term, y=Count, fill=-log10(PValue)), stat='identity')+
coord_flip() + scale_fill_gradient(low="blue", high = "red")+
xlab("") + ylab("") + theme(axis.text.x=element_text(color="black", size=12),
axis.text.y=element_text(color="black", size=12)) +
scale_y_continuous(expand=c(0, 0)) + scale_x_discrete(expand=c(0,0))
dev.off()
GEO差异分析与WGCNA交集
- 选取模块与性状相关性热图中相关系数最大的模块进行分析(准备该模块中的基因)
- 准备GEO差异分析结果(|logFC|>1且p<0.05)
#install.packages("VennDiagram")
library(VennDiagram)
setwd("C:\\Users\\lexb4\\Desktop\\WGCNA\\14.venn")
files=dir() #获取目录下所有文件
files=grep("txt",files,value=T) #提取.txt结尾的文件
geneList=list()
#读取所有txt文件中的基因信息,保存到geneList
for(i in 1:length(files)){
inputFile=files[i]
if(inputFile=="intersect.txt"){next}
rt=read.table(inputFile,header=F)
header=unlist(strsplit(inputFile,"\\.|\\-"))
geneList[[header[1]]]=as.vector(rt[,1])
uniqLength=length(unique(as.vector(rt[,1])))
print(paste(header[1],uniqLength,sep=" "))
}
#绘制venn图
venn.plot=venn.diagram(geneList,filename=NULL,fill=rainbow(length(geneList)))
pdf(file="venn.pdf",width=6,height=6)
grid.draw(venn.plot)
dev.off()
#保存交集基因
intersectGenes=Reduce(intersect,geneList)
write.table(file="intersect.txt",intersectGenes,sep="\t",quote=F,col.names=F,row.names=F)















 663
663











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









