







AbMole(奥默生物)是ChemBridge在中国的唯一官方指定合作伙伴。
由来自山东大学齐鲁医学院的 Ying Zhou,JingJing Ye以及ChunYan Ji等多名研究人员在期刊Adv Sci (Weinh)(IF=15.1)上,共同发表了题为“Silencing of IRF8 Mediated by m6A Modification Promotes the Progression of T-Cell Acute Lymphoblastic Leukemia”的文章。在该文章中,研究人员使用了购自AbMole的FB23-2(M9422)和 Actinomycin D(M4881)两种产品,揭示了IRF8在T细胞急性淋巴细胞白血病(T-ALL)中的抑制作用,并阐明FTO通过调控IRF8的m6A甲基化修饰,从而介导T-ALL发病的具体机制,为T-ALL的研究提供了极具潜力的新策略。

T-ALL是一种侵袭性血液恶性肿瘤,并且预后较差。因此探究T-ALL的发生发展机制,寻找新的研究靶点,对攻克T-ALL具有重要的科学价值。
研究人员通过比较T-ALL患病受试者以及健康人之间的差异表达基因(DEGs),发现IRF8在T-ALL患病受试者中受到异常抑制(图1A-B)。然后通过慢病毒转染,在T-ALL细胞系Molt4和Jurkat中过表达IRF8,并进行CCK8和EdU检测等实验,发现上调IRF8能显著抑制T-ALL细胞的生长(图1C)以及侵袭迁移能力(图1D-E)。
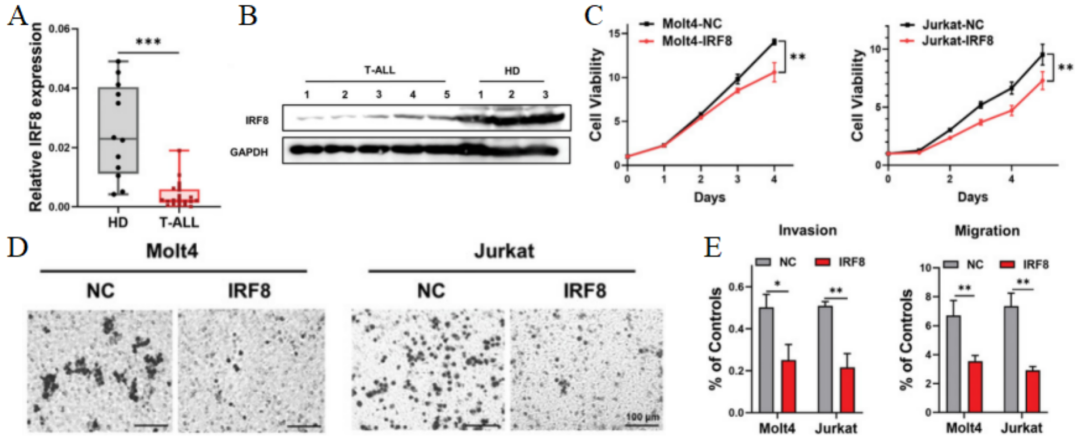
图 1 A. 通过qRT-PCR检测新诊断的T-ALL患病受试者和健康供体骨髓中IRF8的表达。B. 通过免疫印迹法检测新诊断的T-ALL患病受试者和健康供体骨髓中IRF8的表达。C. 通过CCK8试验分析IRF8表达的Molt4细胞、Jurkat细胞和NCs的增殖情况。D. 过表达IRF8的Molt4和Jurkat细胞,以及NC细胞在Matrigel基质膜下的结晶紫染色。E. 过表达IRF8的Molt4细胞和NC的侵袭和迁移能力。
接下来,研究人员通过构建Irf8敲除小鼠进行实验(图2A),结果显示,Irf8-/-组小鼠的T-ALL发展具有高侵袭性,并且潜伏期更短,同时表现出明显的肝脾肿大和股骨苍白,说明存在白细胞增多、红细胞减少和多器官浸润等情况(图2B)。此外,基因敲除小鼠骨髓GFP+细胞中Ki67水平升高,表明敲除Irf8能促进白血病细胞的增殖(图2C),提示Irf8的缺失能与驱动基因协同作用,共同加速T-ALL发展。
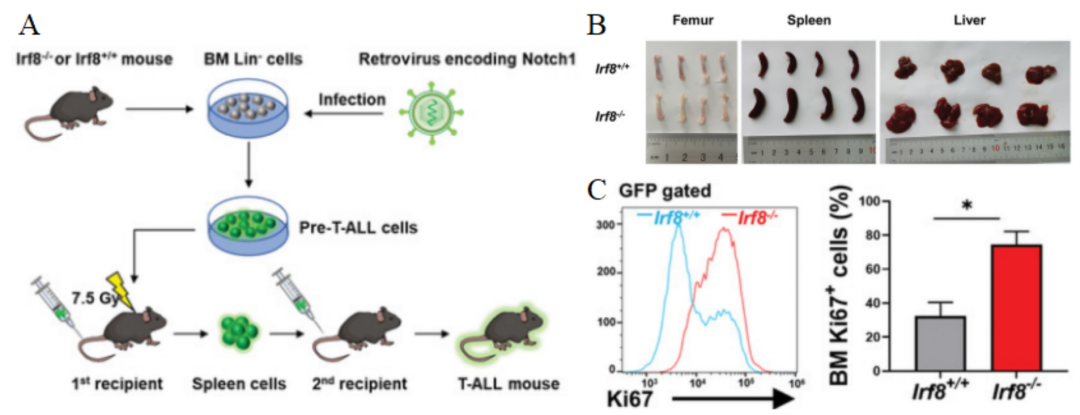
图 2 A. 建立T-ALL小鼠模型的程序。B. 移植了Irf8-/-和Irf8+/+脾细胞的小鼠在移植后两周时的股骨、脾脏和肝脏图像。C. 移植两周后,Irf8-/-组和Irf8+/+组GFP+门控骨髓细胞的Ki67表达情况。
KEGG对DEGs的分析显示,PI3K/AKT信号通路显著富集(图3A)。随后选择PI3K/AKT信号相关DEGs—PIK3R5(图3B)进行进一步研究。免疫印迹(图3C)和CCK8(图3D)结果显示,在IRF8过表达的Molt4细胞中,PIK3R5表达下调,AKT和mTOR的磷酸化水平降低。表明在T-ALL中,IRF8通过PIK3R5负调控PI3K/AKT通路。
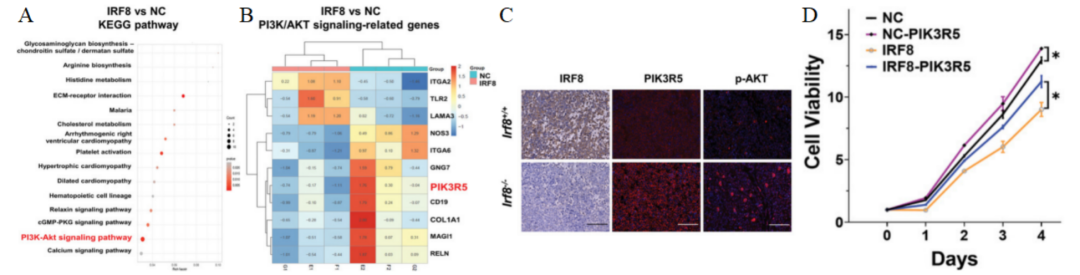
图 3 A. KEGG分析显示,与阴性对照(NC)Molt4细胞相比,RNA-seq检测到IRF8表达的Molt4细胞中PI3K/AKT通路富集。B. 热图显示了IRF8表达的Molt4细胞(E1、F1、G1)和阴性对照(NC)Molt4细胞(E2、F2、G2)中与PI3K/AKT信号相关的DEGs。PIK3R5被确定为DEG。C. Irf8-/-和Irf8+/+ T-ALL小鼠脾脏中IRF8、PIK3R5和p-AKT表达的免疫荧光结果。D. PIK3R5过表达对Molt4-NC和Molt4-IRF8细胞活力的影响。
之后,研究人员使用Jaspar数据库,在PIK3R5的启动子区域发现IRF8的潜在结合位点(图4A)。染色质免疫沉淀(ChIP)显示,IRF8免疫沉淀中的PIK3R5启动子序列显著富集(图4B),表明IRF8可直接识别PIK3R5启动子序列中的结合基序,调控PIK3R5转录。
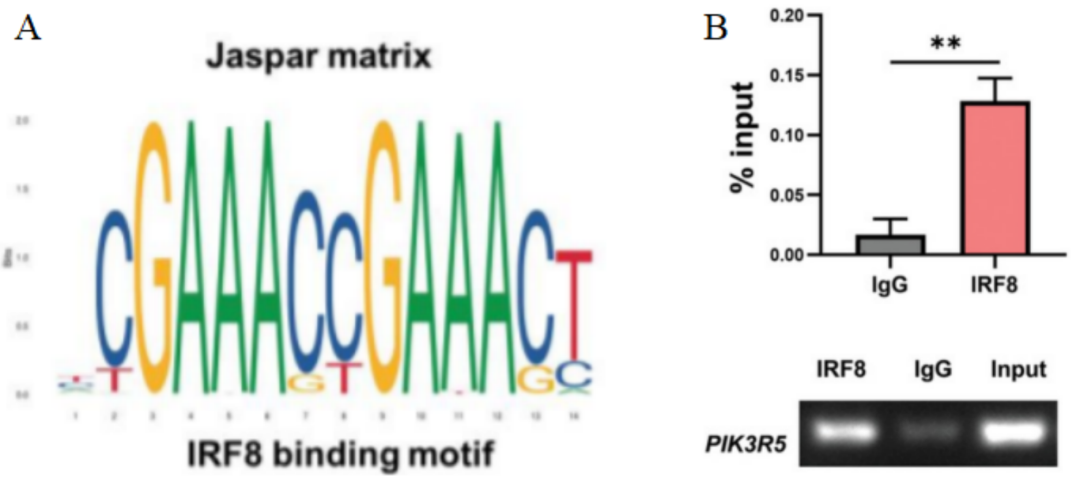
图 4 A. Jaspar数据库预测的PIK3R5启动子区域中可与IRF8结合的潜在基团。B. Molt4细胞中PIK3R5的ChIP‐qPCR分析和相应的电泳图谱。
随后,研究人员对T-ALL数据集GSE13159进行基因组富集分析(GSEA),结果显示T-ALL患病受试者体内各种m6A调控因子发生显著变化,其中以FTO的变化水平最为明显(图5A)。Pearson相关分析表明,在两个独立的T-ALL群体中,FTO和IRF8的表达存在明显的负相关性(图5B)。
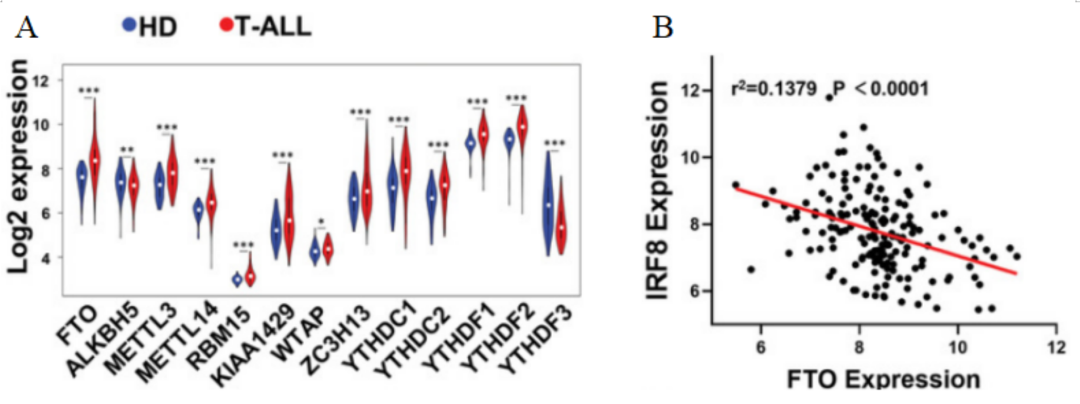
图 5 A. T-ALL患病受试者和健康供体(HDs)骨髓中m6A调节因子的Log2表达值。B. 来自GSE13159的174份T-ALL样本中FTO和IRF8表达水平的相关性,Pearson相关性分析。
接下来,用小发夹RNA(shRNA)慢病毒敲除FTO或使用FB23-2抑制FTO水平进行实验,检测发现,两种方式均会导致IRF8表达水平升高(图6A-B),证明IRF8的表达受到FTO介导的m6A修饰的负调控。
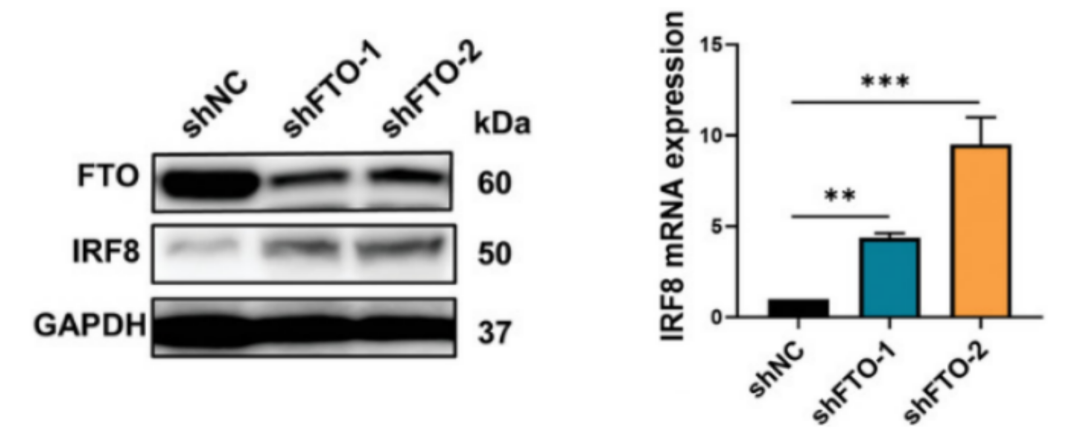
图 6 A. 通过Western印迹检测转染FTO shRNA慢病毒(shFTO-1和shFTO-2)或阴性对照(shNC)的Molt4细胞中FTO和IRF8的水平。B. 转染shFTO-1、shFTO-2或shNC慢病毒的Molt4细胞中IRF8 mRNA水平的qRT-PCR分析。
随后,研究人员使用FB23-2处理Molt4细胞,发现m6A的总体丰度提高(图7A),且IRF8的表达提高2至3倍(图7B)。MeRIP-seq分析显示,FTO失活导致IRF8 3′非翻译区(UTR)的m6A水平明显增加。此外,FTO RIP-seq分析显示,FTO结合峰富集在IRF8 mRNA转录本中,IRF8转录本3′UTR中的轨迹与m6A峰重叠(图7C)。
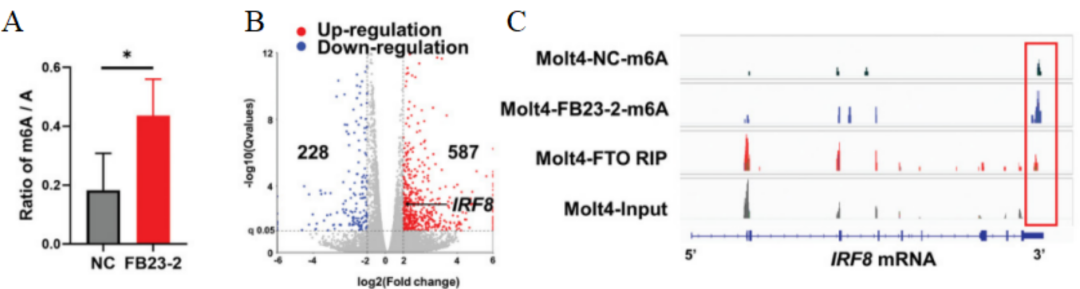
图 7 A. 用2 µm FB23-2或DMSO(NC)培养48小时的Molt4细胞中mRNA的总体m6A丰度,并用LC-MS/MS描述m6A/A的比率。B. DEGs的火山图;IRF8用黑点标出。蓝点:下调基因;红点:上调基因。C. Integrative Genomics Viewer (IGV)追踪描绘用DMSO载体或FB23-2处理的Molt4细胞中IRF8基因的m6A修饰,以及IRF8基因座中的FTO RIP-seq信号和相应的输入信号。
之后设计引物扩增IRF8 3′UTR中包含预测的潜在结合位点的序列(图8A),并进行RIP-qPCR检测,共筛选出五个可能被FTO直接结合的潜在m6A位点(图8B)。随后,将候选m6A位点从A到T分别替换(MUT1至MUT5,图8C),并构建野生型和五个突变型IRF8 3′UTR报告载体。经研究发现,转染WT报告载体细胞的荧光素酶活性明显高于HEK293T-shNC细胞。并且转染MUT1至MUT5报告载体后,可以恢复荧光素酶活性(图8D)。表明IRF8是FTO的一个关键靶点,FTO通过这些m6A位点负调控IRF8 mRNA的表达。随后,研究人员使用FB23-2和Actinomycin D处理Molt4和Jurkat细胞,发现IRF8转录本的半衰期明显延长(图8E)。表明抑制FTO的去甲基化活性可通过促进RNA稳定性和减少RNA降解来恢复IRF8 mRNA的表达。
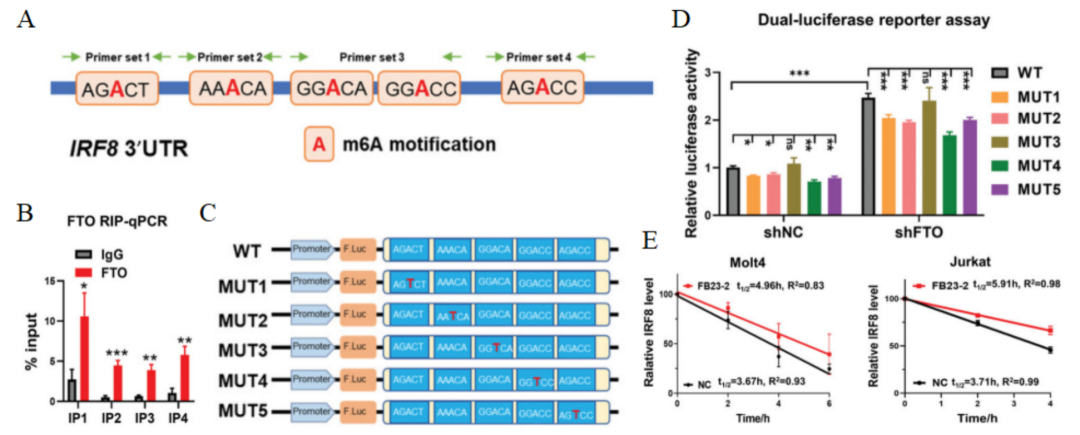
图 8 A. IRF8 3′ UTR中潜在的m6A结合位点(位点1-5)示意图。引物组3(IP3)包括两个位点:位点3和位点4。B. FTO RIP-qPCR分析显示FTO与Molt4细胞中IRF8 mRNA转录本结合。C. 克隆到pMIR-REPORT荧光素酶载体的IRF8 mRNA 3′ UTR野生型(WT)以及m6A识别位点从A核苷酸突变为T核苷酸的五个突变序列(MUT1至MUT5)示意图。D. 野生型IRF8 mRNA 3′ UTR(WT)和突变型IRF8 mRNA 3′ UTR(MUT1至MUT5)报告载体转染有或无FTO敲除的HEK293T细胞的相对荧光素酶活性。E. 用5 µm FB23-2或DMSO(NC)预处理Molt4和Jurkat细胞24小时,并在指定时间用5 µg/mL Actinomycin D培养的IRF8 mRNA降解检测,并与0小时的mRNA水平归一化。
最后研究人员使用FB23-2处理Irf8+/+组和Irf8-/-组的T-ALL小鼠模型(图9),并通过进行流式细胞术,点印迹检测以及免疫印迹等分析。结果表明,抑制FTO的m6A去甲基化酶活性,能恢复T-ALL中IRF8的表达,从而通过PI3K/AKT信号转导抑制T-ALL的发生。

图 9 建立Irf8-/-和Irf8+/+ T-ALL小鼠模型及FB23-2给药时间表(FB23-2:2 mg/kg)。
该研究使用了购自AbMole的FB23-2(M9422)和 ActinomycinD(M4881)两种产品,FB23-2是一种新型FTO小分子抑制剂,可直接与FTO结合,选择性地抑制m6A去甲基化酶的活性,在外显子RNA甲基化靶向研究领域具有巨大的潜力和优势。在该实验中,研究人员多次使用FB23-2抑制FTO活性,进而更好的研究FTO与IRF8表达之间的调控机制。Actinomycin D是一种转录抑制剂,在该实验中用于在 mRNA降解分析实验中抑制转录,避免新转录的mRNA影响实验结果。
研究人员使用FB23-2抑制FTO的活性,发现IRF8的表达水平升高,细胞增殖受到FB23-2剂量依赖性抑制(图10A-B),并可以通过敲除IRF8得到有效缓解。进一步研究发现,FB23-2处理和IRF8敲除之间在细胞活力(图10C)和集落形成活性(图10D)方面具有明显的交互作用,说明IRF8的表达受到FTO介导的m6A修饰的负调控。
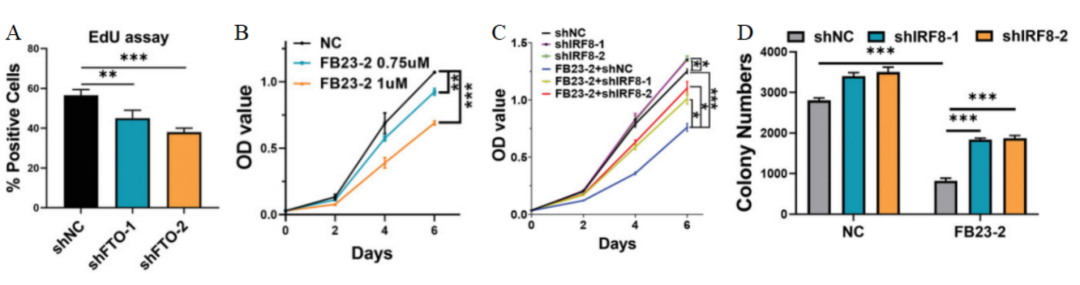
图 10 A. 用不同浓度的FB23-2或DMSO(NC)处理Molt4细胞48小时的IRF8 mRNA水平。B. 用不同浓度的FB23-2或DMSO(NC)处理的Molt4细胞的增殖情况。C. 用shIRF8-1、shIRF8-2或shNC慢病毒转染的Molt4细胞经FB23-2处理后的活力。D. 转染shIRF8-1、shIRF8-2或shNC慢病毒并经1 µm FB23-2处理的Molt4细胞的集落形成能力。
此外,研究人员通过对Irf8+/+组和Irf8-/-组的T-ALL小鼠进行FB23-2处理(图9)。流式细胞术显示,给药FB23-2能显著降低Irf8+/+组骨髓、血液和脾脏中GFP+细胞的比例,以及Ki67的水平(图11A),并且小鼠的存活时间延长(图11B)。说明FB23-2能有效减轻Irf8+/+T-ALL小鼠的白血病负担并延长其生存期。免疫印迹和免疫荧光分析表明,FB23-2能显著增加Irf8+/+组小鼠骨髓和脾脏白血病细胞中IRF8的表达,并抑制PIK3R5的表达和AKT的磷酸化(图11C-D)。因此,研究人员认为FB23-2抑制FTO的m6A去甲基化酶活性,能成功恢复T-ALL中IRF8的表达,并能通过抑制PI3K/AKT信号转导,从而抑制白血病的发生过程。
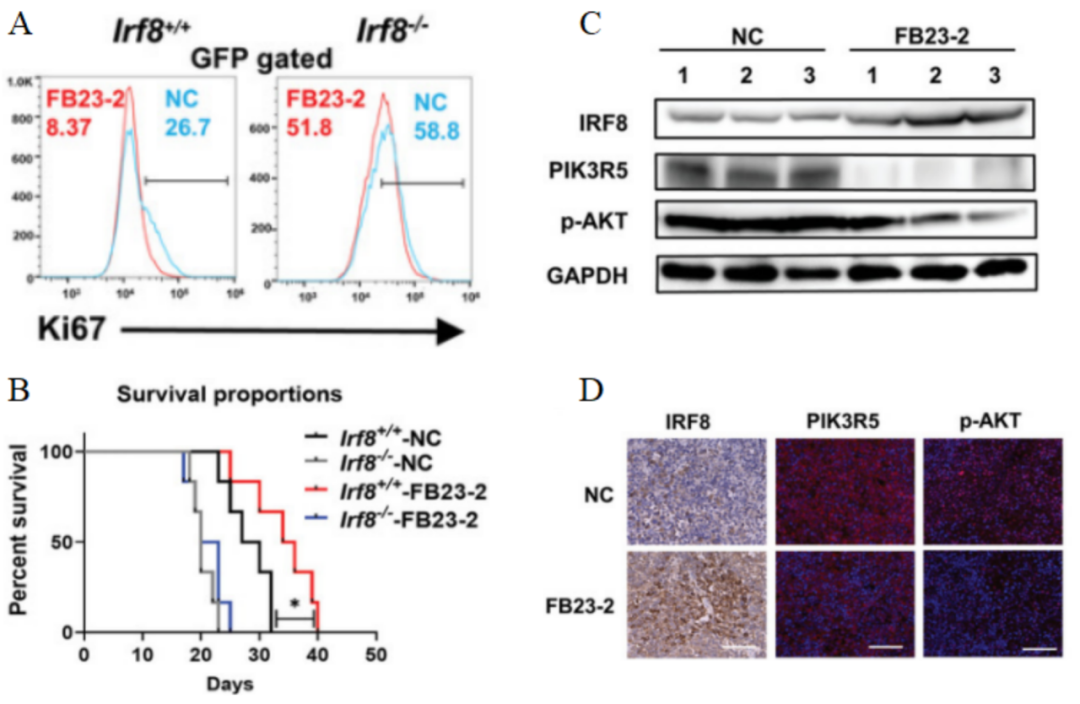
图 11 A. 流式细胞仪测定的GFP+门控骨髓细胞的Ki67表达水平。B. 用FB23-2或DMSO(NC)处理的Irf8-/-组和Irf8+/+组小鼠的存活率。C. 免疫印迹分析FB23-2或DMSO(NC)处理的Irf8+/+T-ALL小鼠骨髓中的IRF8、PIK3R5和p-AKT水平。D. 给药FB23-2或DMSO(NC)后Irf8+/+T-ALL小鼠脾脏中IRF8、PIK3R5和p-AKT表达的免疫荧光图片。
更多内容请访问:AbMole官网(www.abmole.cn)
















 204
204

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









