







1.Introduction
minfi包适用于分析450K和850K的甲基化矩阵。
每个样本在一个矩阵上以两个不同的颜色通道(red、green)进行测量;每个矩阵测量4.5E5个CpG,对每个CpG的甲基化和未甲基化进行度量。
样品之间的DNA甲基化差异可以在单个CpG处,称为差异甲基化位置(DMP),也可以在区域水平上,称为差异甲基化区域(DMR)。
2.minfi object
(1) RGChannelSet : IDAT文件的原始数据,红绿通道。
(2) MethylSet : CpG基因座水平数据,甲基化和未甲基化通道。
(3) RatioSet : Beta和M通道(或之一)。
(4) GenomicMethylSet : 比对到基因组的MethylSet。
(5) GenomicRatioSet : 比对到基因组的RatioSet。

** 3. 准备输入数据**
(1) sample sheet : 一个记录实验设计的csv文件。
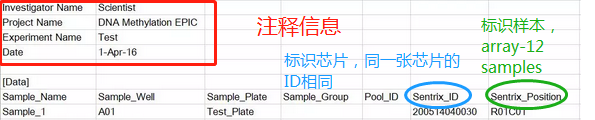
(2)一个样本会有两个.idat文件,是红绿双通道。整理如下目录:

4. 读取输入数据
targets = read.metharray.sheet(baseDirectory,pattern = "SampleSheet.csv")
RGset <- read.metharray.exp(targets=targets)
pd <- pData(RGset)
5.质量过滤
(1) 过滤可信度较低的探针:p > 0.01。通过detectP 这个函数计算探针对应的p值。
probeP = detectionP(RGset)
keep = apply(probeP,1,function(x) {all(x < 0.01)})
RGset = RGset[keep,]
过滤质量差的样本:样本所有探针P的均值>0.05
keep = apply(probeP,2,mean) < 0.05
RGset = RGset[,keep]
(2) 获取芯片注释信息
Mset = preprocessRaw(RGset)
Gset = mapToGenome(Mset)
annotation = getAnnotation(Gset)
(3) 过滤覆盖了snp位点的探针
获取snp信息:snps = getSnpInfo(Gset)

CpG_rs是指这个CpG位点同时也是一个snp位点。
SBE_r指的是CpG位点下游的第一个碱基是snp位点。
Probe_rs 指的是探针区域覆盖到的snp位点。
因此要过滤掉:Gset = dropLociWithSnps(Gset,snps = c("CpG","SBE","CpG"))
(4) 过滤掉位于性染色体上的探针
sex_probe = rownames(annotation) [annotation$chr %in% c("chrX","chrY")]
keep = !(featureNames(Gset) %in% sex_probe)
GRset = Gset[keep,]
6.预处理
去除噪声:通过control探针的信号强度来校正CpG探针的信号强度。
归一化:I型探针和II 型探针之间的差异、技术重复beta值分布的差异
(1) preprocessIllumina:Genomestudio 软件
Mset.illumina = preprocessIllumina(RGset,bg.correct = TRUE,normalize = "controls")
Beta.illumina <- getBeta(Mset.illumina)
(2) preprocessSWAN : SWAN算法
Mset.swan = preprocessSWAN(RGset)
Beta.swan = getBeta(Mset.swan)
(3) preprocessQuantile
Mset.quantile = preprocessQuantile(RGset,
fixOutliers = TRUE,
removeBadSamples = TRUE,
badSampleCutoff = 10.5,
quantileNormalize = TRUE,
stratified = TRUE,
mergeManifest = FALSE,
sex = NULL)
Beta.quantile <- getBeta(Mset.quantile)
(4) preprocessNoob : noob算法。
Mset.noob = preprocessNoob(RGset)
Beta.noob = getBeta(Mset.noob)
(5) preprocessFunnorm : noob算法降低噪声,functional normalization算法归一化。
Mset.funnorm = preprocessFunnorm(RGset)
Beta.funnorm = getBeta(Mset.funnorm)
7.差异点位DMP分析
beta = getBeta(GRset)
group = pd$Sample_Group
dmp = dmpFinder(beta,pheno = group,type = "categorical")
pheno参数用于指定样本的分组情况。
type参数用于指定分组类型:case/control不同的实验处理就是categorical;分组是连续型变量就是continuous。
8.差异区域DMR分析
group = pd$Sample_Group
design_matrix = model.martix(~group)
dmrs = bumphunter(GRset,
design = design_matrix,
cutoff = 0.2,
B = 0,
type = "Beta")















 245
245











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









