







软件名称 | 主要功能 |
XRD数据分析工具 | |
XPS数据分析工具 | |
XRD数据分析 | |
拉曼光谱分析 | |
核磁共振NMR数据处理 | |
科研图像处理工具 | |
XPS数据分析 | |
观察生物分子微观结构 | |
纳米颗粒粒度分析软件 | |
原子水平晶体结构可视化 | |
文献管理软件 | |
数据分析工具 | |
三维建模和渲染软件 | |
电镜图像分析软件 | |
晶体分子结构图绘制软件 | |
非线性拟合软件 | |
晶体结构解析软件 | |
热化学分析软件 | |
红外数据分析软件 | |
科研绘图软件 | |
分子材料计算模拟软件 | |
XPS和AES数据分析软件 | |
Xcalibur | 质谱数据分析软件 |
Mercury | 观察晶体结构软件 |
Nanoscope Analysis | AFM数据处理软件 |
Image Pro Plus | 图像分析软件 |
ZSimpWin | 阻抗谱拟合软件 |
Masshunter | 质谱采集分析软件 |
Cytoscape | 生物信息分析软件 |
CrysTBox | TEM数据分析软件 |
Demeter | 同步辐射XAFS软件 |
TA advantage | 热重分析软件 |
Gaussian16 | 量子化学计算软件 |
TA Universal Analysis | 热分析软件 |
目录
软件下载
软件介绍
安装教程
使用教程
软件下载
[安装环境配置]:
1.Ubuntu 20.04.1 LTS
2.GeForce RTX 3080
3.Driver Version: 470
4.CUDA Version: 11.1
[下载地址]:https://ambermd.org/GetAmber.php
软件介绍
Amber(Assisted Model Building with Energy Refinement)是一个专为分子建模、分子动力学模拟和计算化学设计的库。它提供了一整套工具,用于研究生物大分子,如蛋白质、核酸和小分子。Amber项目由一组开发者维护,主要包括University of California, San Francisco (UCSF)的研究团队。
软件特点:
1. 提供全面的分子动力学模拟功能,包括能量计算、最小化和动力学模拟。
2. 支持多种力场,允许用户选择适合特定分子的模型。
3. 集成了多种分析工具,可以处理和分析分子动力学产生的数据。
4. 具有与其他计算化学软件(如GROMACS和CHARMM)的兼容性。
5. 社区活跃,提供丰富的文档和用户支持。
科研服务器推荐:
分子动力学模拟通常需要处理大量的粒子相互作用和复杂的物理建模,同时还需要高速并行计算来支持大规模模拟。因此,强大的计算性能、高效的内存管理和优化的并行架构是不可或缺的。
以下是一种推荐配置,适合主流分子动力学模拟场景,同时支持按需定制以满足特定需求。
安装教程
Amber 的安装分为多个步骤,确保根据系统环境和要求进行适当的设置。以下是安装过程的基本步骤:
基本安装指南
1.系统要求:确保您的计算机满足最低硬件和软件要求。
2.获取源代码:从 Amber 官方网站下载最新版本的源代码。
3.解压源代码:将下载的源代码解压到指定目录。
4.配置环境:根据您的计算机系统配置适当的环境变量和软件依赖。
5.编译程序:使用适合您系统的编译器编译 Amber 程序。
6.测试安装:运行提供的测试程序以确保安装成功。
本安装教程以Amber20为例子,教程部分转载自公众号【大科研小分享】。
也可按照官方教程手册进行安装,后台回复【Amber 2020】获取pdf文件。

一、安装相关依赖
这里直接根据官网给出的代码运行即可
apt -y update
apt -y install tcsh make \
gcc gfortran \
flex bison patch \
bc xorg-dev libbz2-dev wget如果要安装并行程序,须运行以下代码
apt -y install openmpi-bin libopenmpi-dev openssh-client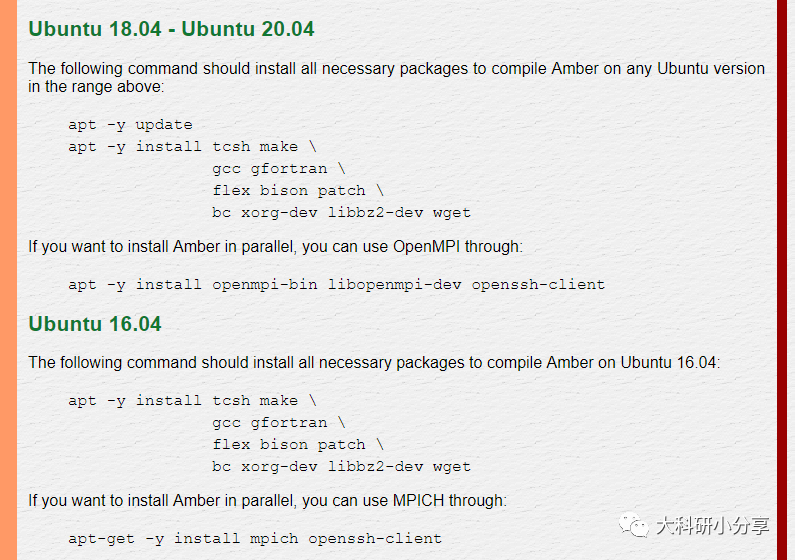
注意:对于新安装系统的机子,上面没有readline包,因此也需要安装,在ubuntu系统下,readline的包名称命为libreadline-dev,因此还需安装这个包,否则在编译时会报错
sudo apt-get install libreadline6-dev二、cmake安装
之前已经提到过Amber20版本通过cmake安装,因此需要安装CMake
关于CMake的安装很简单,有几种方法可以轻松地在Linux上安装CMake。
1. 如果使用Anaconda,则可以使用conda install cmake来获取最新的cmake,
2. 如果系统支持snap,则可以通过sudo snap install cmake--classic将cmake作为snap安装。
3. 或者从官网下载tar.gz文件安装(https://cmake.org/download/)
三、正式安装
1. 首先将Amber20和AmberTools放在同一文件夹,并分别解压。
tar xvf AmberTools21.tar.bz2
tar xvf Amber20.tar.bz2注意,必须通过以上命令进行安装,不要使用linux图形界面的提取。
解压完成后会出现amber20_src的文件夹

2. 通过cd切换到amber20_src/build文件夹,然后运行CMake脚本进行编译。因为小编这里已经编译过一次,所以出现的文件有点多,第一次进入build文件夹时应该只有四个文件
cd amber20_src/build
sudo ./run_cmake
3. 编译完成后运行以下代码进行安装,这里-j 20代表使用20核进行编译,加快速度。安装完成后,在amber20_src文件夹所处位置会出现Amber20
sudo make -j 20
sudo make install4. 修改环境变量
在home文件夹修改bashrc文件,在最后一行添加以下代码,读者可根据自己安装的路径进行设置。然后在终端输入source ~/.bashrc即可

如果这里不清楚,可参见小编以前的推文
AMBER20 GPU和并行版本
上述步骤只能安装amber的串行版本,若想安装amber的并行,GPU,NCCL多GPU通讯版本,则需要修改run_cmake文件进行再次编译。
1. 首先我们需要安装并行和GPU版本所需的openMPI和cuda。安装方法可查看小编之前的推文。注意:安装到任意的文件夹都行,只要设置好环境变量就行
2. 再次切换到amber20_src/build文件夹,用写字板打开run_cmake文件,将-DMPI=FLASE,-DCUDA=FLASE改为-DMPI=TRUE,-DCUDA=TRUE
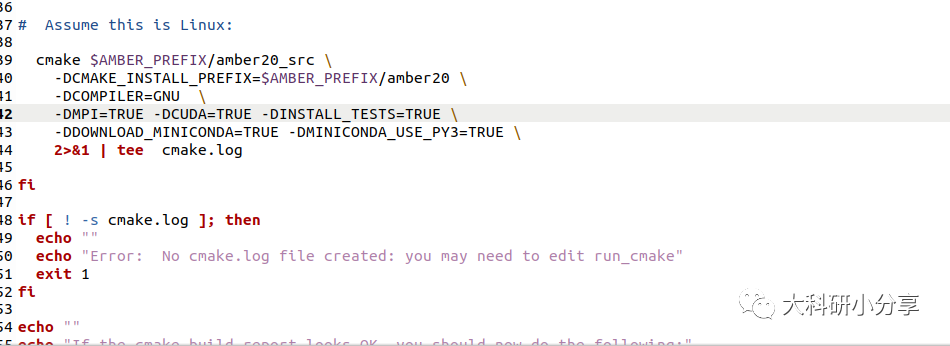
修改完成后,保存退出,再次运行
sudo ./run_cmake运行完成后,可查看编译结果,发现cuda和mpi都已经完成编译,然后直接安装即可
sudo make -j 20
sudo make install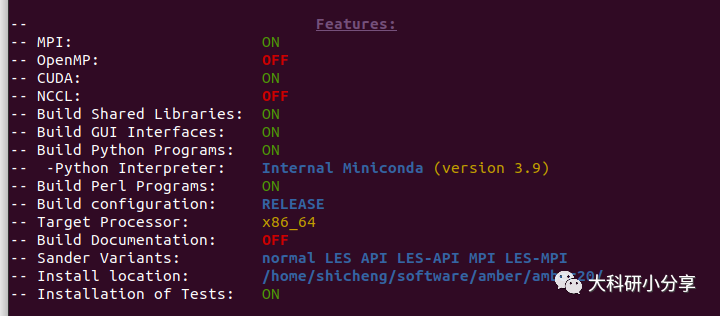
注意:小编有看到网上有些教程在安装amber20时先安装串行版本,然后在安装并行版本,再安装GPU版本,再安装GPU并行版本,然而,小编在安装时直接将需要安装的版本改成TURE即可,不需要反复编译。
参考来源
https://blog.csdn.net/qq_33953882/article/details/113995531
总的来说,就是想要装哪个版本,直接把相关位置改成TRUE即可
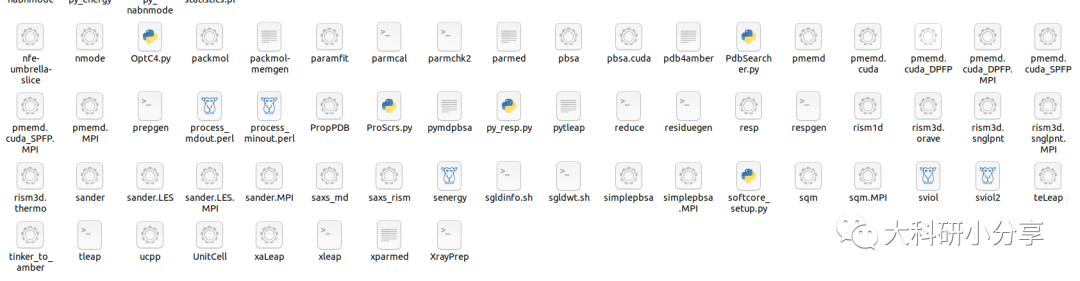
安装完成后,会出现并行版本的pmemd.MPI,GPU版本的pmemd.cuda,混合精度浮点版本pmemd.cuda_SPFP与支持双精度浮点版本pmemd.cuda_DPFP,GPU并行版本的pmemd.cuda_SPFP.MPI与pmemd.cuda_DPFP.MPI,其中pmemd.cuda_SPFP.MPI即为之前版本的pmemd.cuda.MPI。
若想安装NCCL,则将run_cmake中相应位置改为-DNCCL=TRUE即可
应用场景
1.药物设计
Amber 软件套件提供了强大的分子动力学模拟工具,能够帮助研究人员研究药物分子与靶标(如蛋白质或核酸)之间的相互作用。通过 Amber 的分子动力学模拟和自由能计算功能,研究人员可以预测药物分子的结合模式、计算结合自由能,并优化药物分子的结构。这些功能在药物筛选和先导化合物优化中具有重要作用。
2.蛋白质工程
Amber 的力场(如 ff14SB 和 ff19SB)专为蛋白质模拟设计,能够高效地模拟蛋白质的动力学行为。研究人员可以利用 Amber 研究蛋白质的折叠过程、构象变化以及与其他分子的相互作用,从而为蛋白质设计和功能优化提供理论支持。此外,Amber 还支持对膜蛋白的模拟,这对于研究嵌膜蛋白质的功能和设计新型蛋白质工具具有重要意义。
3.生物物理研究
Amber 是研究生物分子动力学和相互作用的强大工具。通过 Amber 的分子动力学模拟,研究人员可以深入了解生物分子的行为特征,例如蛋白质-核酸复合物的形成机制、膜蛋白与脂质双层的相互作用,以及分子在溶液中的动力学特征。Amber 的模拟结果可以为实验设计提供指导,并帮助验证实验数据。
4.分子行为与相互作用研究
Amber 提供了多种工具(如 sander 和 pmemd)用于分子动力学模拟和能量最小化,能够帮助研究人员探索分子间的相互作用和动力学特征。例如,Amber 可以模拟蛋白质-配体、蛋白质-核酸或蛋白质-蛋白质复合物的形成过程,并通过轨迹分析工具(如 cpptraj)深入研究这些相互作用的细节。
5.理论研究与实验指导
Amber 的高效模拟能力使其成为理论研究的重要工具。研究人员可以使用 Amber 模拟复杂的生物分子体系,并通过分析模拟结果验证理论模型。此外,Amber 的模拟结果可以为实验设计提供重要参考,例如预测分子构象变化、识别关键相互作用位点以及优化实验条件。
温馨提示:分享的所有软件,均由互联网中的资源整理所得,仅限学习交流,切勿商用,侵删!
—The end—
更多资源请关注元素魔方科研服务

















 717
717

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









