










GROMACS 分子动力学模拟 蛋白-配体分子动力学模拟
简介
在本篇博客中,我将介绍如何使用 GROMACS 进行以下操作:
- 分子对接 提取最佳结合能的对接结果,然后抽取配体。
- 安装Linux(Ubuntu | CentOS)版本的Grace、VMD1.9.4 、Gromacs2024.1 CUDA支持的 GPU加速版;
- 对膜蛋白进行分子动力学模拟
- VMD查看系统结构文件图像(md.gro)
- VMD查看轨迹文件图像(md.trr)
- VMD查看转换后的轨迹图像(traj.pdb)
- VMD查看能量最小化或平衡阶段的轨迹(em.gro / npt.gro / nvt.gro)
- VMD分析文件——将 RMSF 数据映射到蛋白质的 B 因子字段,显示结构中柔性较高和较低的区域。
- Grace 查看蛋白质(或其他分子)随时间变化的均方根位移(RMSD);
- Grace 查看分子中每个原子(或特定组分)随时间的均方根波动(RMSF);
- Grace 查看能量相关数据,通常包括势能、动能、总能量等(energy.xvg);
- Grace 查看模拟过程中压力的变化(pressure.xvg);
- Grace 查看模拟过程中的温度数据(temperature.xvg);
- Grace 查看体系的密度变化数据(density.xvg);
这里给出几篇有助于新手了解、安装Gromacs需要知道什么 准备什么的友情链接:
Gromacs 分子动力学 远程安装介绍 全网最详细的Gromacs安装前说明 该怎么选择合适的安装方式 Windows直接可用的Gromacs(预编译版)有什么危害?Gromacs安装需要准备什么?
【分子动力学】 分子动力学新手入门:一文读懂GROMACS使用全流程,轻松开启模拟之旅! GROMACS初学者了解资料 GROMACS安装之前的准备 windows如何安装Gromacs
本文适用于对分子动力学模拟有基础了解的读者,并重点关注 蛋白-配体复合物 和 膜蛋白 的模拟过程和分析结果展示。
环境准备
本次软件与工具
- GROMACS2024.1 CUDA 支持的GPU加速版
- 操作系统:Ubuntu22.04
- 绘图工具、蛋白查看: Python 的 Matplotlib 库、Grace、VMD、Pymol3.1 开源版
准备数据文件 —— 分子对接 AutoDock 、 Vina(软件安装可与我联系)
SailVina 使用教程 Autodock Vina分子对接全套整合软件 MGLTools闪退 作用力分析 MOE 薛定谔 Gromacs 全网最全分子对接教程
AutoDock对接流程 AutoDockTools AutoDock对接总流程 AutoDock Vina
分子对接软件
Linux系统安装AutoDockTools、AutoGrid和AutoDock并实现分子对接 分子对接软件 CentOs
Ubuntu系统适用(详细讲解)
AutoDock与Vina的区别 如何选择AutoDOCK与Vina对接 AutoDock对接系列 run
AutoGrid出现[Error 2]从而得不到.map文件 AutoDock安装闪退
- 蛋白-配体复合物的初始结构文件(
test.pdb) - 这里声明一下,对接出来的蛋白,进行模拟是需要拆分受体和配体的。纯对接物,直接给gmx 力场使用是会报错的哦!
分子对接过程后,筛选出结合能最低(即结合稳定性最高)的对接构象,然后将该构象的蛋白和配体整合成一个文件,保存为包含两者的 .pdb 格式文件;会用到Pymol可视化对接结果。
检查对接产生的结果文件是否符合Gromacs模拟的要求
- 检查PDB 文件的命名规则是否符合 GROMACS 使用的力场标准。
发现缺失原子:则需要修复缺失的原子
方法 1:使用 PyMOL 打开 test.pdb 文件:进行调整。接着使用 Mutagenesis 工具补全残基缺失的原子。方法 2:使用 SwissSidechain SwissSidechain 是一个在线工具,可修复非标准残基和补全原子。
- 使用 pdb4amber 工具规范化 PDB 文件
使用标准化后的文件才能运行 gmx pdb2gmx
使用Groamcs处理模拟之前PDB文件,并进行MD模拟 核心命令 思路顺序
这里展示下我的配体:
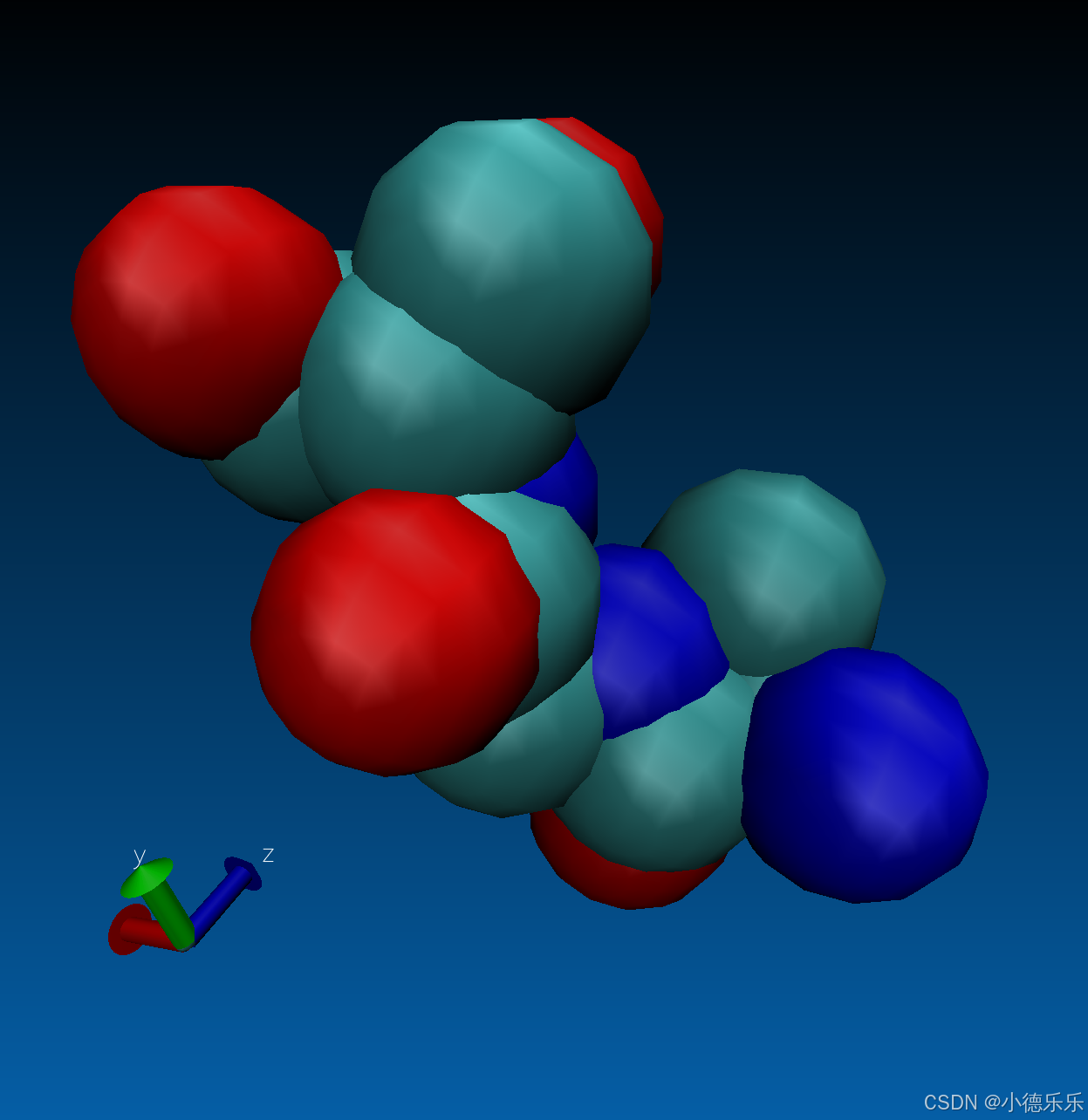
1. gmx pdb2gmx:将 PDB 文件转化为 GROMACS 格式,生成初始的结构和拓扑文件
2. gmx editconf: 定义盒子的大小和形状,设置为立方体盒子,边界距离 1.0 nm
3. gmx solvate: 在盒子中添加溶剂分子,使用 spc216.gro 作为溶剂配置
4. gmx grompp: 准备离子添加的输入文件
5. gmx genion:添加离子并中和体系,使用 NA 和 CL 作为离子类型
6. gmx grompp: 准备能量最小化的输入文件
这里需要自己查询官网文档:准备minim.mdp文件
这里提供一个例子:
integrator = steep
emtol = 1000.0
emstep = 0.01
nsteps = 50000
cutoff-scheme = Verlet
ns_type = grid
coulombtype = PME
rcoulomb = 1.0
rvdw = 1.0
pbc = xyz
7. gmx mdrun: 执行能量最小化,启用GPU 加速
8. gmx energy: 提取能量最小化的势能数据
9. gmx grompp: 准备 NVT 平衡的输入文件
10. gmx mdrun: 执行 NVT 平衡模拟 启用GPU 加速
11. gmx energy: 提取 NVT 平衡过程中的温度数据
12. gmx grompp :准备 NPT 平衡的输入文件
13. gmx mdrun :执行 NPT 平衡模拟 启用GPU 加速
14. gmx energy : 提取 NPT 平衡过程中的压力和密度数据
gmx energy -f npt.edr -o pressure.xvg
gmx energy -f npt.edr -o density.xvg
15. gmx grompp :准备生产模拟的输入文件
这里需要自己查询官网文档:准备md.mdp文件
这里提供一个例子:
integrator = md
nsteps = 5000000 ; 模拟 10 ns
dt = 0.002 ; 时间步长 2 fs
nstxout = 5000 ; 每 10 ps 输出坐标
nstvout = 5000 ; 每 10 ps 输出速度
nstenergy = 5000 ; 每 10 ps 输出能量
nstlog = 5000 ; 每 10 ps 输出日志
continuation = yes
constraint_algorithm = lincs
constraints = all-bonds
cutoff-scheme = Verlet
tcoupl = V-rescale
tc-grps = System
tau_t = 0.1
ref_t = 300
pcoupl = Parrinello-Rahman
pcoupltype = isotropic
tau_p = 2.0
ref_p = 1.0
compressibility = 4.5e-5
16. gmx mdrun :执行最终的生产模拟 启用GPU 加速
gmx mdrun -deffnm md -gpu_id 0
分析并提取模拟结果
1. 提取轨迹为 PDB 格式,用于可视化
gmx trjconv -s md.tpr -f md.trr -o traj.pdb
2. 计算 RMSD,分析体系稳定性
gmx rms -s md.tpr -f md.trr -o rmsd.xvg
3. 计算 RMSF,分析分子柔性
gmx rmsf -s md.tpr -f md.trr -o rmsf.xvg
4. 提取能量数据,分析体系的总能量变化
gmx energy -f md.edr -o energy.xvg
VMD可视化和结果展示
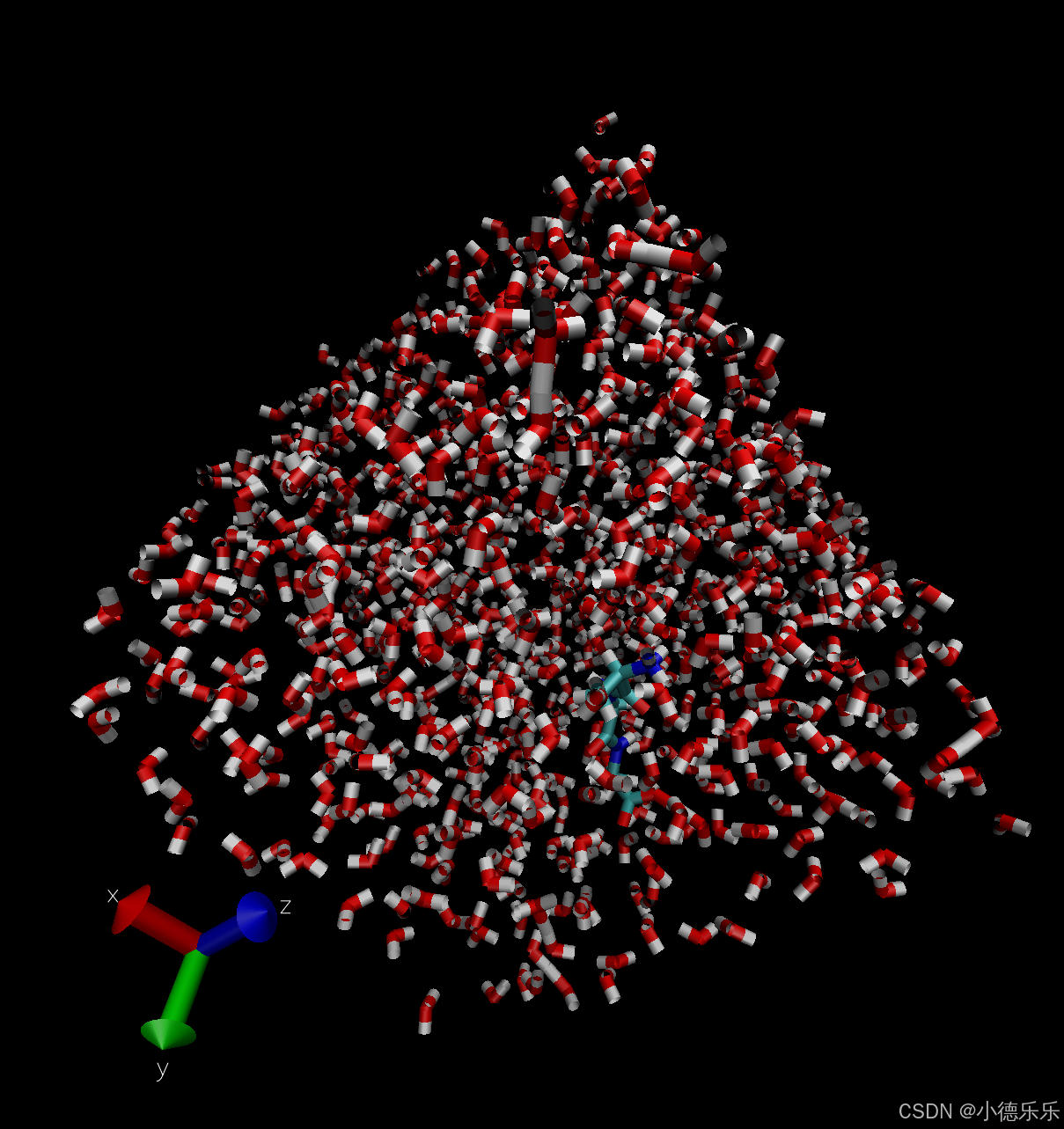
- 系统结构文件
文件:md.gro
内容:生产模拟结束时的最终系统结构,包括蛋白、溶剂、离子等。
用途:查看最终结构,检查模拟是否合理,如溶剂分布、蛋白质构象等。

- 轨迹文件
文件:md.trr
内容:生产模拟的完整轨迹数据,包括所有时间帧的原子坐标。
用途:以动画形式观察分子运动,如蛋白质折叠、构象变化、溶剂和离子的运动等。
这里实际是一个可播放的动画,描述轨迹运动,这里只做截图展示。

- 能量最小化或平衡阶段的轨迹
文件:em.gro / npt.gro / nvt.gro
内容:模拟不同阶段(能量最小化、NVT、NPT 平衡)的最终结构。
用途:比较不同阶段的系统结构变化,例如溶剂填充是否完整、蛋白质构象是否合理。
em.gro:

4.观察溶剂分布和离子排布:
加载 md.gro 和 md.trr,观察溶剂与蛋白的相互作用。

Grace可视化和结果展示
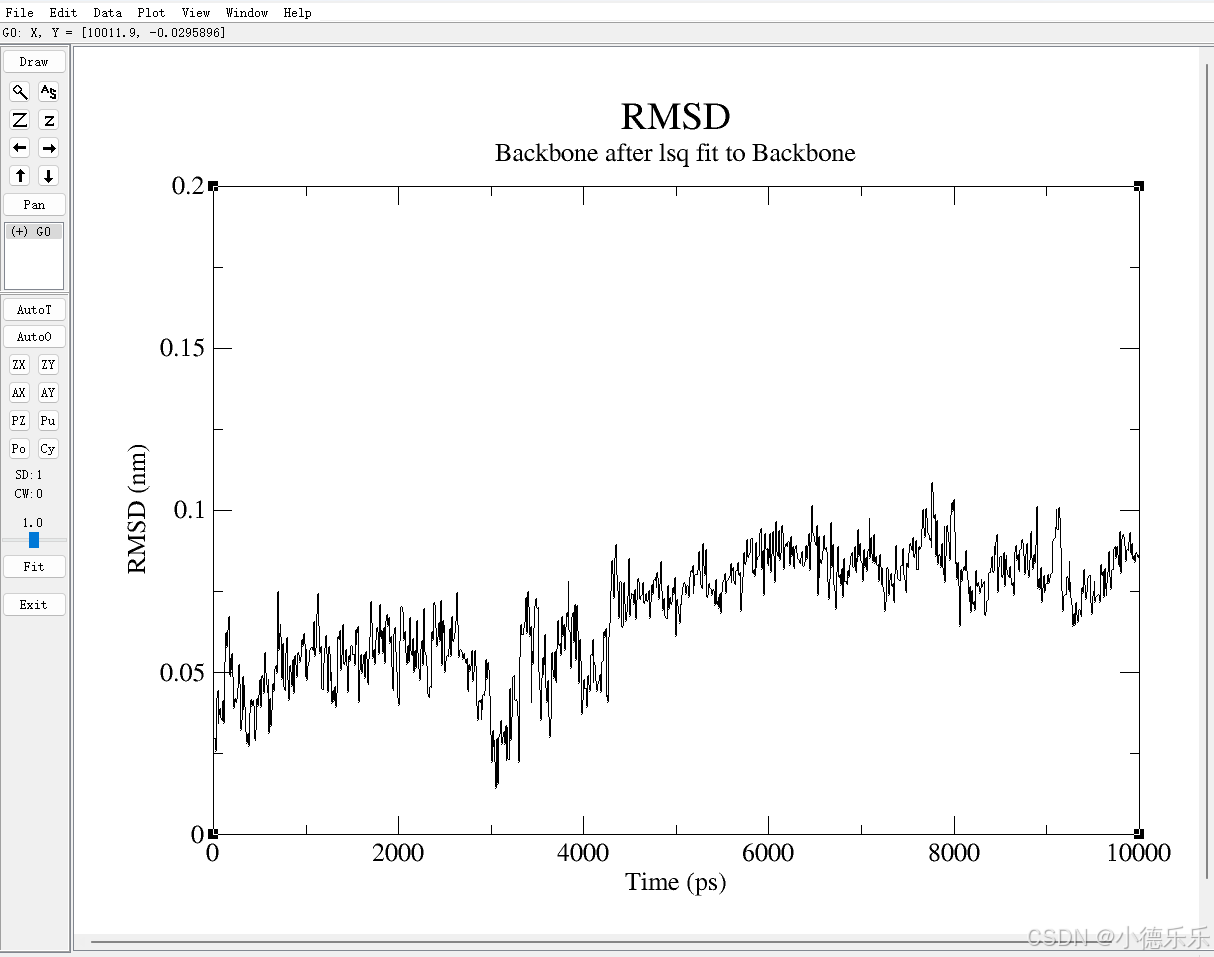
- RMSD (rmsd.xvg)
文件内容:包含了蛋白质(或其他分子)随时间变化的均方根位移(RMSD)。
图形展示:会显示模拟过程中分子结构的稳定性。RMSD 越小表示结构越稳定,波动较大时则可能表示系统正在经历过渡或不稳定状态。
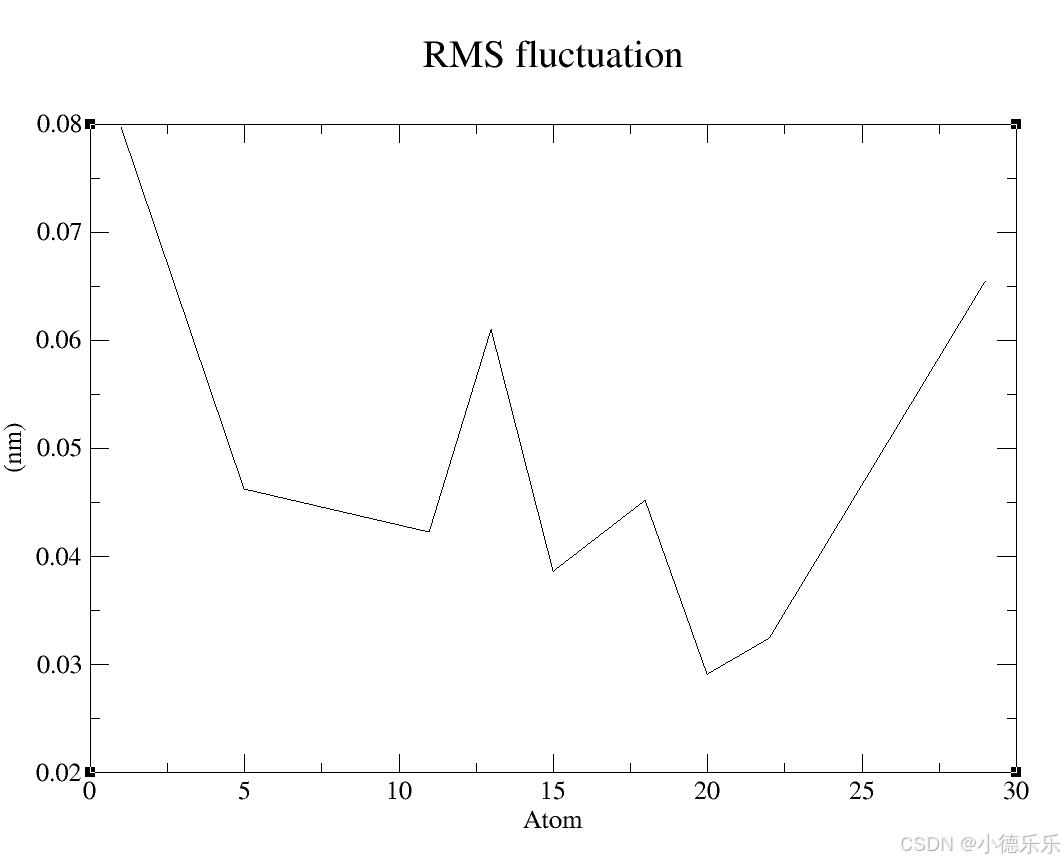
2.RMSF (rmsf.xvg)
文件内容:包含了分子中每个原子(或特定组分)随时间的均方根波动(RMSF)。
图形展示:显示各个原子或残基的波动程度,波动较大的区域通常代表灵活或有较大运动范围的区域,如侧链、环结构等。
-
energy.xvg: 文件内容:包含了能量相关数据,通常包括势能、动能、总能量等。
图形展示:帮助你监控模拟过程中的能量变化,判断系统是否达到平衡态。例如,能量波动较大可能表示系统尚未平衡。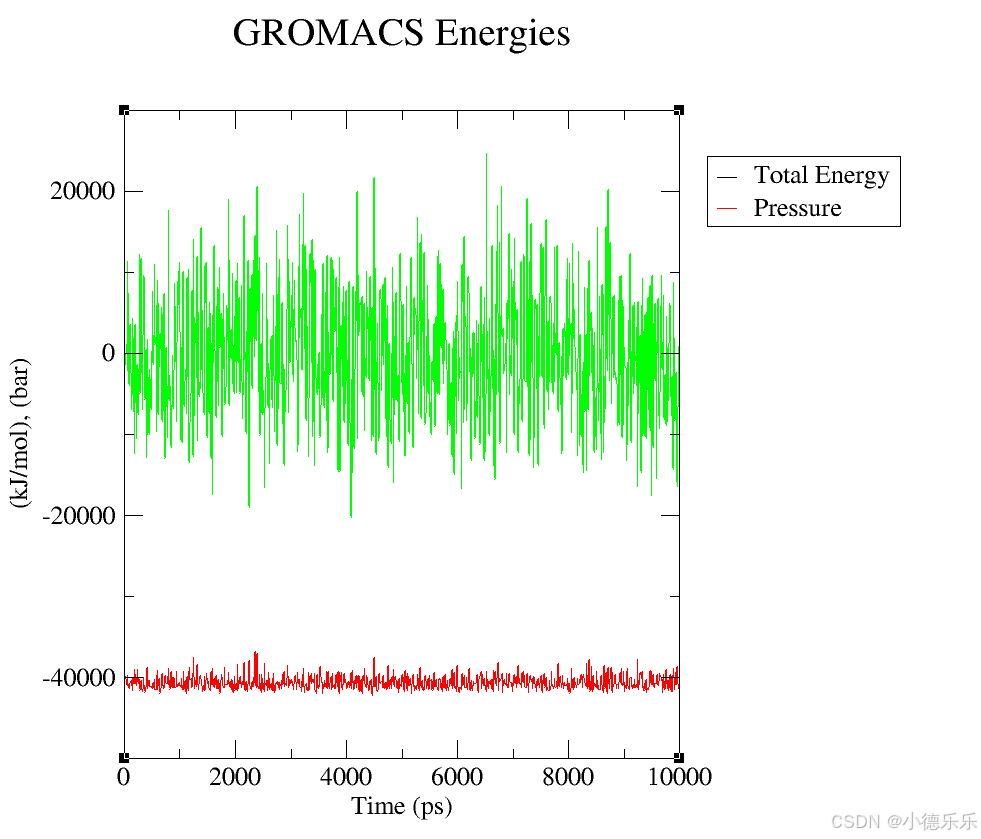
-
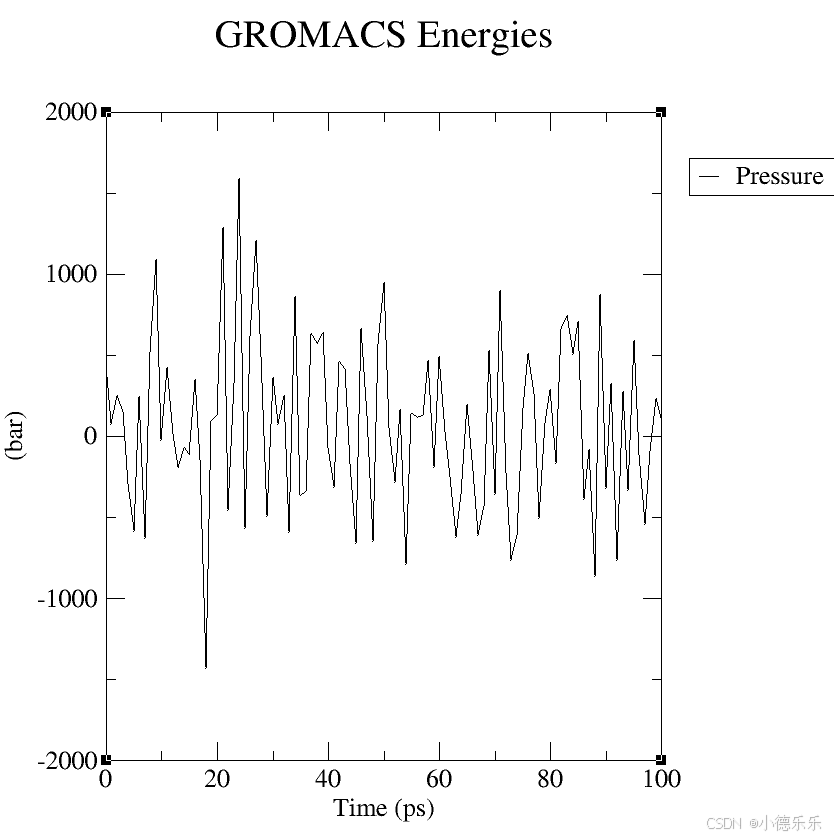
pressure.xvg: 文件内容:包含了模拟过程中压力的变化。
图形展示:你可以查看压力如何随着时间变化,通常用于分析体系是否达到了平衡状态。如果压力波动较大,可能意味着模拟还没有达到稳定状态。
-
temperature.xvg: 文件内容:包含了模拟过程中的温度数据。
图形展示:用来查看模拟过程中温度的变化。如果温度波动较大,可能表示系统没有达到热平衡。
-
density.xvg: 文件内容:包含了体系的密度变化数据。
图形展示:显示模拟过程中的密度变化。密度的变化可能与溶剂化、压缩、扩张等有关。
结论
- 模拟结果显示膜蛋白在水溶液中的稳定性良好,其结构变化与柔性特性符合生物学特性。
- 能量、温度、压力等参数的稳定表明体系构建合理,模拟流程成功完成。
- 使用 RMSD 和 RMSF等分析指标能够有效评估膜蛋白的构象变化,为后续的功能研究提供支持。
- 可视化工具(如 VMD 和Grace)直观展示了膜蛋白的动力学行为和轨迹数据,增强了对体系的理解。
通过本次模拟,展示了 GROMACS 在膜蛋白分子动力学模拟中的完整流程,并获得了一套可靠的分析方法和结果展示。如果有更多深入的研究需求,可以与我联系一起探究。
与我联系——解决AutoDock对接报错以及闪退,提供对接教学服务。
PC端电脑通过
点击PC端分子对接软件合集——“能看到某宝对应的分子对接软件商品!!!。
手机淘宝通过:
点击手淘分子对接软件合集 “——能看到某宝对应的分子对接软件商品!!!



















 765
765

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









