







来自生信技能树课程
目录
目录
一、常用方法
library(tinyarray)
find_anno("GPL代码",install = T)#GPL代码通常在数据资料内可找到
#>- [1] `library(xxxxx.db);ids <- toTable(xxxxxxx)` and `ids <- AnnoProbe::idmap('GPL代码')` are both available
#2选1运行代码即可下载GPL注释文件当上述代码运行结果为:no annotation packages avliable,please use `ids <- AnnoProbe::idmap('XXXGPLXXX')` if you get error by this code ,please try different `type` parameters
二、不可行,以下4选1下载注释文件
1.Bioconductor R包(最常用)
#找到GPL码
#GPL平台与R包的对应关系:http://www.bio-info-trainee.com/1399.html
#安装并加载相应的R包(此处以为例"hgu133plus2.db")
if(!require(hgu133plus2.db))BiocManager::install("hgu133plus2.db")
library(hgu133plus2.db)
ls("package:hgu133plus2.db")#列出某R包内有哪些函数/数据
#用toTable()处理探针注释号(xxxSYMBOL)且不能加引号(此处为“hgu133plus2SYMBOL”);
ids <- toTable(hgu133plus2SYMBOL)
head(ids)2.读取GPL网页的表格文件,按列取子集
(1)下载GPL文件到工作目录
- 网站下载:GEO数据库官网,跟查找下载GSE相同。
(以GPL570为例):https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570 - GEOquery包下载:getGEO("GSExxxxx",getGPL =T) ——有时会连接超时无法下载
- get_gpl_txt {tinyarray}也可以下载:get_gpl_txt("GPLxxxx")(很大下载很慢)
(2)继续
#读取表格(可应用函数众多,视情况而定)
#表格读取参数、文件列名不统一,活学活用,有的表格里没有symbol列,也有的GPL平台没有提供注释表格
b = read.delim("GPL570.txt",
check.names = F,
comment.char = "#")
#check.names——是否将列名里的特殊符号转换为下划线
#comment.char = "#"——#开头的行是注释行,不作为表格正式内容
#"skip ="为跳过开头几行
colnames(b)
ids2 = b[,c("ID","Gene Symbol")]
#为避免数据覆盖,命名为ids2,自己运行时最后要运行ids=ids2以实现代码数据衔接
colnames(ids2) = c("probe_id","symbol")
#为使与之后代码兼容更改列名,此处列名不可动!!
#检查ids2数据有误异常并进行相应处理(以下为常见问题举例)
#查看ids2可见symbol有空值,即有的探针没有对应的symbol,要将其删除
k1 = ids2$symbol!=""
table(k1)
#有的探针对应多个symbol(非特异探针),将其删除
k2 = !str_detect(ids2$symbol,"///")
table(k2)
ids2 = ids2[ k1 & k2,]
#ids = ids23.官网下载注释文件并读取(很有可能不能用)
http://www.affymetrix.com/support/technical/byproduct.affx?product=hg-u133-plus
4.自主注释
https://mp.weixin.qq.com/s/mrtjpN8yDKUdCSvSUuUwcA
三、探针注释数据异常的处理
1.基因(symbol)有空值
即有的探针没有对应的symbol,要将其删除
ids=ids$symbol!=""#保留非空值
#ids<-na.omit(ids)也可以:查找不适用的记录;或删除不适用的记录2.基因有缺失值
any(is.na(ids))#检查有缺失值(NA)
ids<-na.omit(ids)#na.omit()查找不适用的记录;删除不适用的记录3.一个探针(id)对应多个基因(symbol)
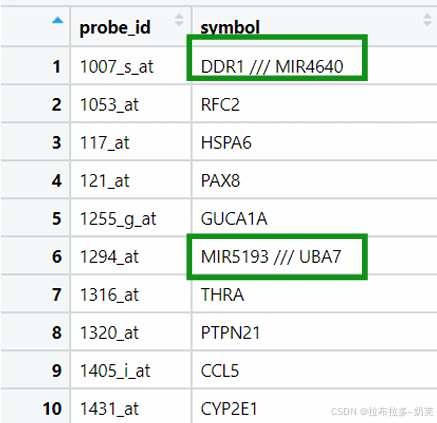
(1).删除该行
ids= !str_detect(ids$symbol,"///")#删除含有“///”的那一行(2).保留分隔符号前面的第一个基因
ids0<- apply(ids,1,
function(x){paste(x[1],
str_split(x[2],'///',simplify=T),
sep = "...")
})
x = tibble(unlist(ids0))
colnames(x) <- "ttt"
ids <- separate(x,ttt,c("probe_id","symbol"),sep = "\\...")
dim(ids)
第一个发现非常简单,在使用merge( )或trans_array( )匹配时,会剔除更多的基因。第二个方法,会保留更多基因。
3.多个探针(id)对应一个基因(symbol)
当多个探针对应可1个基因时,不能说是基因表达了多次,所以要去重
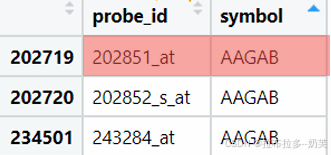
以下方法各有道理,无所谓好坏
(1).随机去重
ids = ids[!duplicated(ids$symbol),]#ids按照symbol去重(2).保留行和最大/行平均值最大的探针(从众原则)
#保留最大值
exp2 = exp[ids$probe_id,]
identical(ids$probe_id,rownames(exp2))
ids = ids[order(rowSums(exp2),decreasing = T),]#rowSums()为行和,rowMeans()为行平均值
ids = ids[!duplicated(ids$symbol),]
nrow(ids)
# 如果进行差异分析,拿这个ids去inner_join得到deg进行差异分析(3).取多个探针的平均值
#求平均值
exp3 = exp[ids$probe_id,]
rownames(exp3) = ids$symbol
exp3[1:4,1:4]
exp4 = limma::avereps(exp3)#压缩微阵列数据对象,以便用平均值替换阵列内重复探针的值
# 此时拿到的exp4已经是一个基因为行名的表达矩阵,直接差异分析,不再需要inner_join

















 3万+
3万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









