#merge进行多组seuratobject数据合并
# 合并多个Seurat对象
gpy.big <- merge(COEA,
y = c(xxxx, xxxx, xxxx),
add.cell.ids = c("xxxxx", "xxxx", "xxx", "xxxx"),
project = "gpy")
#Harmony去批次/基因染色/umap图/差异表达基因和表达式热图
gpy.big <- NormalizeData(gpy.big, normalization.method = "LogNormalize",
scale.factor = 10000) ##后面是函数的默认值
#根据前5000个高变基因及进行UMAP
gpy.big <- FindVariableFeatures(gpy.big, selection.method = "vst", nfeatures = 5000) ##默认返回nfeatures =2000
all.genes <- rownames(gpy.big)
gpy.big <- ScaleData(gpy.big, features = all.genes)#中心化
#PCA之后harmony去批次
gpy.big <- RunPCA(gpy.big, features = VariableFeatures(object = gpy.big), verbose = F)
library(harmony)
gpy.big <- gpy.big %>% RunHarmony("orig.ident", plot_covergence = T)
##去批次以后umap
gpy.big<- gpy.big %>%
RunUMAP(reduction = "harmony", dims = 1:30) %>%
FindNeighbors(reduction = "harmony", dims = 1:30) %>%
FindClusters(resolution = 0.1) %>%
identity()
# 创建一个空的pdf文件,用于保存图形输出
pdf("D:/AXOEA.pdf")
##画图按orig.ident
DimPlot(gpy.big, reduction = "umap",group.by = "orig.ident")
##画图按群
DimPlot(gpy.big, reduction = "umap",label = T)
###寻找差异表达基因(findallmarkers)
##devtools::install_github("immunogenomics/presto")
gpy.big.markers <- FindAllMarkers(gpy.big, only.pos = TRUE)
gpy.big.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1)
##染色
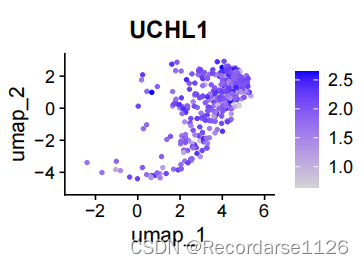
FeaturePlot(gpy.big, features = c("POU5F1", "SOX2", "DNMT3B", "POU3F1"))
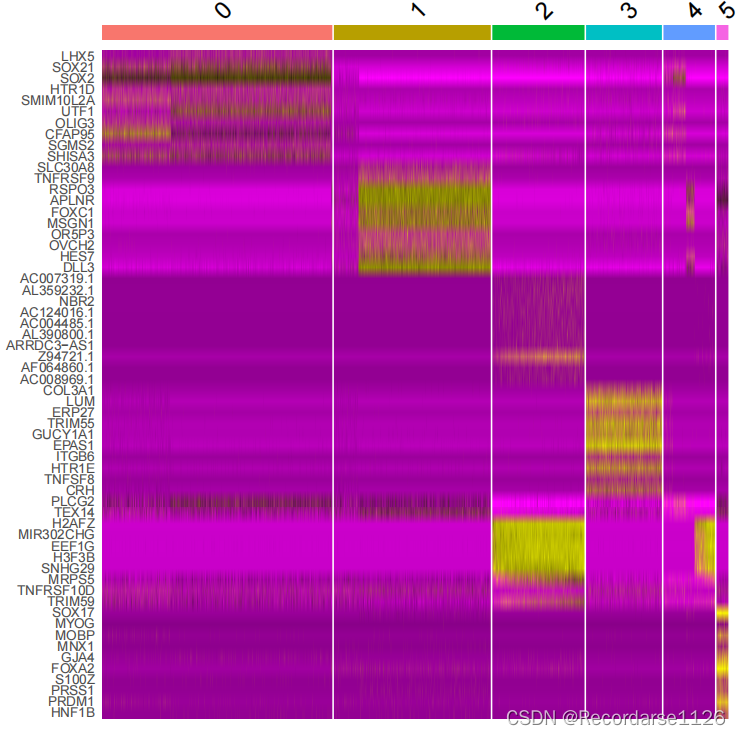
##表达式热图
gpy.big.markers %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 10) %>%
ungroup() -> top10
DoHeatmap(gpy.big, features = top10$gene) + NoLegend()
# 关闭pdf设备,保存图形输出
dev.off()






















 552
552











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









