






一、Cellranger以及参考基因组的下载(linux)
截至目前10×Genomics的官网显示Cellranger的最新版为“Cell Ranger (tar.gz compression) - 7.2.0” ,我们首先在Xshell执行下列代码之一下载Linux 64-bit的安装包:
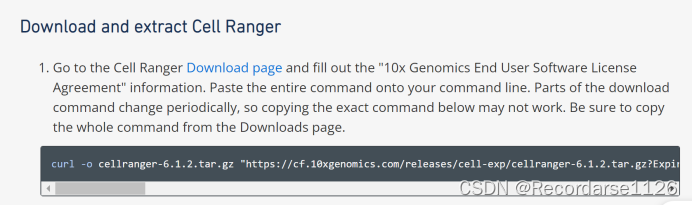
10x官网有教程,可以一步步来,但是我后面下载了miniconda,activate了一个Cellranger环境,方便我后面使用,以上blog不展示此内容,也可以mkdir一个cellapps文件夹下(新手建议)。
安装miniconda:
wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh参考链接:
Linux下conda安装及使用 - 简书 (jianshu.com)
Day3-单细胞数据fastq及cellranger - 简书 (jianshu.com)
遇到错误:例如直接在官网复制这段代码下载6.1.2版本,就会显示下载成功,但一直解压不成功等错误
错误显示:
yulab1@nuca:~/XiaoDi/cellapps$ tar -zxvf cellranger-6.1.2.tar.gz
gzip: stdin: not in gzip format
tar: Child returned status 1
tar: Error is not recoverable: exiting now
解决方法:下载命令的某些部分会定期更改,因此复制官网上面的确切命令可能不起作用。确保从Download page界面复制整个命令:
https://www.10xgenomics.com/support/software/cell-ranger/downloads#download-links
遇到错误:明明已经保存了环境,每次打开cellranger总是找不到命令:(环境总是添加不上,师姐也失败,可能是没有权限)
错误显示:command not found

解决方案:
export PATH=/home/yulab1/XiaoDi/apps/cellranger-7.2.0:$PATH
每次登都临时开一个cellranger的环境
添加路径(shell中输入)——添加失败:
pwd
echo 'export PATH=/home/yulab1/XiaoDi/apps/cellranger-7.2.0:$PATH ~/.bashrc
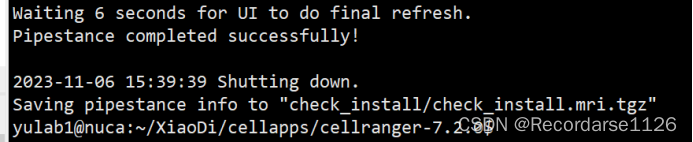
source ~/.bashr二、按照官网的程序,在xshell中测试是否安装成功,显示以下则安装成功:
三、对人的参考基因组和注释进行下载和解压(ensembl官网)
注意人和小鼠不使用toplevel的序列文件:(错误)
curl -O https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.toplevel.fa.gz使用此参考基因组:(正确)
curl -O https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa.gz原因:需要注意的是,大部分物种我们需要下载toplevel的序列文件,但是对于人和小鼠这类有单倍型信息的基因组,我们需要下载primary_assembly的序列。将下载好的文件传到linux主机上。
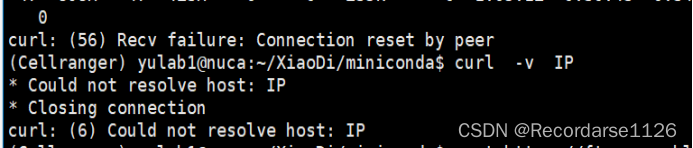
错误显示:Curl使用错误,好像是代理ip问题,我先使用了wget这个命令下载
下载注释:
curl -O https://ftp.ensembl.org/pub/release-110/gtf/homo_sapiens/Homo_sapiens.GRCh38.110.chr.gtf.gz
解压文件:gzip -d Homo_sapiens.GRCh38.dna.toplevel.fa.gz(因为不是tar.gz后缀文件所以不能使用tar命令)
四、过滤注释文件
方法一:
cellranger mkgtf Homo_sapiens.GRCh38.110.chr.gtf Homo_sapiens.GRCh38.110.chr.filtered.gtf --attribute=gene_biotype:protein_coding(运行后也会出现过滤文件,但是进行下一步生成参考基因文件夹就会失败)
方法二:使用以下代码(成功)
cellranger mkgtf \
Homo_sapiens.GRCh38.110.chr.gtf \
Homo_sapiens.GRCh38.110.chr.filtered.gtf \
--attribute=gene_biotype:protein_coding \
--attribute=gene_biotype:lincRNA \
--attribute=gene_biotype:antisense \
--attribute=gene_biotype:miRNA \
--attribute=gene_biotype:IG_LV_gene \
--attribute=gene_biotype:IG_V_gene \
--attribute=gene_biotype:IG_V_pseudogene \
--attribute=gene_biotype:IG_D_gene \
--attribute=gene_biotype:IG_J_gene \
--attribute=gene_biotype:IG_J_pseudogene \
--attribute=gene_biotype:IG_C_gene \
--attribute=gene_biotype:IG_C_pseudogene \
--attribute=gene_biotype:TR_V_gene \
--attribute=gene_biotype:TR_V_pseudogene \
--attribute=gene_biotype:TR_D_gene \
--attribute=gene_biotype:TR_J_gene \
--attribute=gene_biotype:TR_J_pseudogene \
--attribute=gene_biotype:TR_C_gene \五、生成参考基因组文件夹,在服务器中需要1-2h,用nohup命令挂起(发现错误)
在Xshell输入:
nohup cellranger mkref --genome=Homo_sapiens --fasta=Homo_sapiens.GRCh38.dna.toplevel.fa --tered.gtf &错误显示:
nohup报错 nohup: ignoring input and appending output to ‘nohup.out’
参考链接:
nohup报错 nohup: ignoring input and appending output to ‘nohup.out’_centos_我吃西红柿11-华为云开发者联盟
改正代码:
nohup cellranger mkref --genome=Homo_sapiens --fasta=Homo_sapiens.GRCh38.dna.toplevel.fa --tered.gtf > /dev/null 2> /dev/null &
错误显示:以上流程跑出来会说缺少文件
六、不挂nohup命令生成参考基因组
尝试一(错误):
cellranger mkref \
--genome=Homo_sapiens \
--fasta=Homo_sapiens.GRCh38.dna.toplevel.fa \
--genes=Homo_sapiens.GRCh38.110.chr.filtered.gtf && \
--ref-version=3.0.0错误显示:运行成功了,但是又缺少文件,我也不知道为什么
尝试二(失败):
cellranger mkref --genome=Homo_sapiens.GRCh38.110 \
--fasta= Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa \
--genes= Homo_sapiens.GRCh38.110.chr.filtered.gtf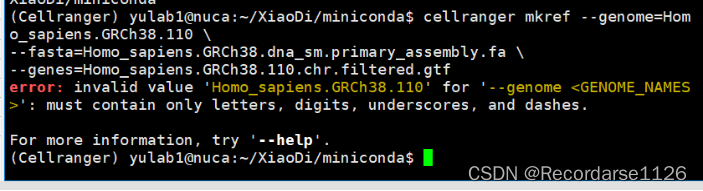
错误显示1:表示 '--genome'<GENOME_NAMES>:只能包含字母、数字、下划线和破折号。
错误显示2:因为等号后面多了空格,导致输入失败
尝试三:(成功)正确代码(跑一个1h左右)
cellranger mkref --genome=Homo_sapiens \
--fasta=Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa \
--genes=Homo_sapiens.GRCh38.110.chr.filtered.gtf 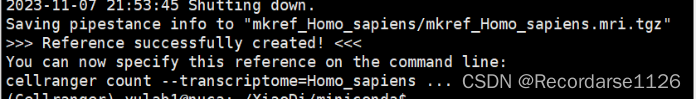
六、cellranger count比对和计数分析


Sample Name命名是CO-EA(但是一般横杠会识别不了,建议改成下划线CO_EA)
改正以后如下:
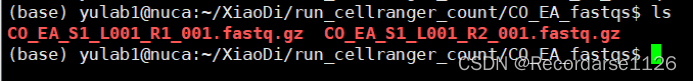
注意新版本的内含子的默认选项(减少测序浪费):
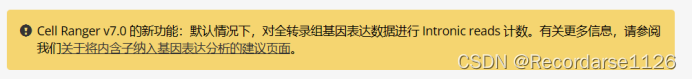


cellranger count分析代码
(1)不含内含子(默认true):
cellranger count --id=nointrons_CO_EA_run_cellranger_count \
--fastqs=/home/yulab1/XiaoDi/run_cellranger_count/CO_EA_fastqs \
--sample=CO_EA \
--transcriptome=/home/yulab1/XiaoDi/miniconda/Homo_sapiens \
--include-introns=false (2)默认含内含子的代码:
cellranger count --id=CO_ET_A_run_cellranger_count \
--fastqs=/home/yulab1/XiaoDi/run_cellranger_count/CO_ET_A_fastqs \
--sample=CO_ET_A \
--transcriptome=/home/yulab1/XiaoDi/miniconda/Homo_sapiens参考设置:
--id:设置输出文件夹路径名称
--fastqs:设置包含目标FASTQ文件的文件夹路径
--sample:如果包含多个样本,指定要分析的样本名 (非必须)
--transcriptome:设置参考基因组文件路径linux基本操作:
创建文件夹:
mkdir CO_ET_AC_fastqs复制文件夹:
cp -r /home/yulab1/XiaoDi/co_culture_sc/Cleandata/CO-ET-A/CO-ET-A_S1_L001_R2_001.fastq.gz /home/yulab1/XiaoDi/run_cellranger_count/CO-ET-A/修改文件名称:
mv CO-ET-A CO_ET_A_fastqs七、cell ranger结果详细解读
关于web_summary.html
Estimated number of cells - 样本测到的细胞数
Mean reads per cell - 每个细胞测到的平均reads
Median genes per cell - 每个细胞基因数的中位数
Sequencing中:
Number of reads - 测到的总read数目
Valid barcodes - UMI校正后匹配的UMI数量
Sequencing saturation:测序饱和度,一般60-80%比较合适(阈值范围可以适当调整,但是高于70%/80%左右绝对OK)
如果测到的细胞数多,但是每个细胞里面的平均reads数少,那么饱和度就不高,反之,饱和度高。但也不是越高越好,背后原理是抽样的原理,到达80%左右就可以代表整个样本。















 1402
1402











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









