







1.数据集 Datasets
(1)Davis
Comprehensive analysis of kinase inhibitor selectivity @ Nat. Biotechnol.2011
442蛋白-68配体,30056对相互作用
dissociation constant (Kd) →
p
K
d
=
−
1
o
g
10
(
K
d
1
e
9
)
pK_d = -1og_{10}(\frac{K_d} {1e9})
pKd=−1og10(1e9Kd)
(2)KIBA
Making sense of large-scale kinase inhibitor bioactivitydata sets: a comparative and integrative analysis @J. Chem. Inf. Model2014
467靶点-52498药物
过滤:至少10对相互作用 → 229蛋白 - 2111药物,118254对相互作用
KIBA(Ki,Kd,IC50) → 构建KIBA分数:Simboost: a read-across approach for predicting drug–target binding affinities using gradient boosting machines.@J. Cheminform.2017
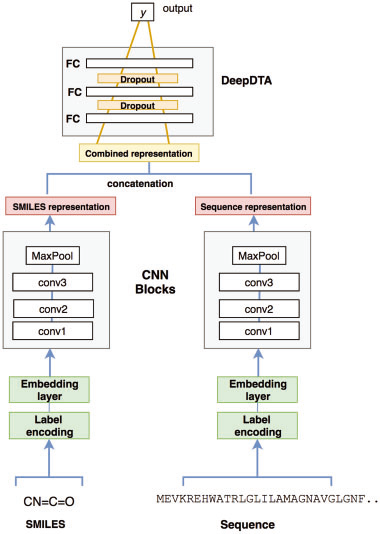
2. 输入表示
(1)SMILE
eg. [C N = C = O] = [1 3 63 1 63 5]
64 labels 固定最大长度 85 (Davis) 100 (KIBA)← 基于分布,cover 80% 蛋白,90%化合物
> > >maximum length 被截断,短序列用0填充
(2) 蛋白序列
Label encoding
25 categories 固定最大长度1200(Davis) 1000(KIBA)
3. 模型
激活函数:ReLU
g
(
x
)
=
m
a
x
(
0
,
x
)
g(x)=max(0,x)
g(x)=max(0,x)
回归任务→ MSE损失函数
M S E = 1 n ∑ i = 1 n ( P i − Y i ) 2 MSE = \frac 1n \sum_{i=1}^n(P_i -Y_i)^2 MSE=n1i=1∑n(Pi−Yi)2
P 是预测向量, Y是真实值向量, n 是样本数量
训练100 个周期,mini-batch size = 256
优化算法 Adam 默认学习率 0.001
使用 Keras Embedding layer ,用128维 dense向量 表示符号
Davis 输入(85,128)和(1200,128)
KIBA 输入 (100,128)和(1000,128)
Concordance Index (CI)测量模型表现,与KronRLS 和 SimBoost 比较
4.实验与结果
(1)Baselines
Kron-RLS的目的是 最小化下面函数,f 是预测函数
J
(
f
)
=
∑
i
=
1
m
(
y
i
−
f
(
x
i
)
)
2
+
λ
∣
∣
f
∣
∣
k
2
J(f) = \sum_{i=1}^m(y_i - f(x_i))^2 + \lambda||f||_k^2
J(f)=i=1∑m(yi−f(xi))2+λ∣∣f∣∣k2
∣
∣
f
∣
∣
k
2
||f||_k^2
∣∣f∣∣k2是 f 的范数,与 核函数k 有关;
λ
>
0
\lambda >0
λ>0 是正则超参数,由用户定义
J
(
f
)
J(f)
J(f) 的最小值可以定义为:
当
f
(
x
)
=
∑
i
=
1
m
a
i
k
(
x
,
x
i
)
f(x) = \sum_{i=1}^ma_ik(x,x_i)
f(x)=i=1∑maik(x,xi) 时
k
k
k是核函数,
J
(
f
)
J(f)
J(f)取得最小。
为了表示化合物,用相似性矩阵表示,由Pubchem structure clustering server(Pubchem Sim)@http://pubchem.ncbi.nlm.nih.gov
对于蛋白 用Smith-Waterman 算法去构建蛋白相似性矩阵
SimBoost
药物,靶点, 药物-靶点对 构建特征
这些特征 喂到 监督学习方法(梯度提升回归树)
任意药物-靶点对
d
t
i
dt_i
dti, 预测的结合亲和力分数
y
i
ˉ
\bar{y_i}
yiˉ 表示为:
y
i
ˉ
=
θ
(
d
t
i
)
=
∑
m
=
1
M
f
m
(
d
t
i
)
,
f
m
∈
F
\bar{y_i} = \theta(dt_i) = \sum_{m=1}^Mf_m(dt_i), f_m \in F
yiˉ=θ(dti)=m=1∑Mfm(dti),fm∈F
M 表示回归树的数量,F表示所有可能的树的空间
正则化目标函数 去学习树 f m f_m fm的参数:
R ( θ ) = ∑ i l ( y i , y i ˉ ) + ∑ m α ( f m ) R(\theta) = \sum_il(y_i,\bar{y_i}) + \sum_m\alpha(f_m) R(θ)=i∑l(yi,yiˉ)+m∑α(fm)
l
l
l是损失函数:测量实际亲和力
y
i
y_i
yi与预测值
y
i
ˉ
\bar{y_i}
yiˉ的差异,
α
\alpha
α是控制模型复杂度的调整参数
为了表示化合物,用相似性矩阵表示,由Pubchem structure clustering server(Pubchem Sim)@http://pubchem.ncbi.nlm.nih.gov
对于蛋白 用Smith-Waterman 算法去构建蛋白相似性矩阵
(2)评价方法
Concordance Index (CI)
C I = 1 Z ∑ δ i > δ j h ( b i − b j ) CI = \frac 1Z \sum_{\delta_i > \delta_j}h(b_i -b_j) CI=Z1δi>δj∑h(bi−bj)
where
b
i
b_i
bi is the prediction value for the larger affinity
δ
i
\delta_i
δi,
b
j
b_j
bj is the prediction value for the smaller affinity
δ
j
\delta_j
δj,
Z
Z
Z is a normalization constant,
h
(
x
)
h(x)
h(x) 是阶梯函数
h
(
x
)
=
{
1
if
x>0
0.5
if
x=0
0
if
x<0
h(x) = \begin{cases}1\quad \text {if \textcolor{orange}{x>0}} \\ 0.5\quad \text{if \textcolor{orange}{x=0}}\\ 0 \quad\text{if \textcolor{orange}{x<0}} \end{cases}
h(x)=⎩
⎨
⎧1if x>00.5if x=00if x<0
MSE
(3)实验设置
数据平分6份,5-fold 交叉验证
确定3 个超参数:过滤器数量(蛋白与化合物相同)
化合物的过滤器尺寸长度,蛋白过滤器尺寸的长度
在验证集上最好的平均CI分数的超参数组合被选为最好的组合去model the test set.
先大范围选,在微调超参数,eg.确定过滤器数量:搜索[16,32,64,128,512], 如果 16最好,范围更新为[4,8,16,20]
使用 r m 2 r_m^2 rm2 和 A U P R AUPR AUPR评价所获模型在测试集上的表现
参考文献:Bioinformatics, 34, 2018, i821–i829
doi: 10.1093/bioinformatics/bty593

















 1264
1264

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









