







在上篇文章中我们描述了miRNA的基本分析流程,本文中我们将对miRNA分析流程中每一个步骤的输出文件进行解释。
软件安装
在snakemake流程中可以使用conda去管理需要的软件,详细的命令使用见: Running Snakemake in conda env, 下面介绍其基本用法:
查看snakemake中conda的环境
snakemake --directory .test --list-conda-envs
结果显示如下
environment container location
../workflow/envs/env.yaml .snakemake/conda/369d9fb0f93ac536da2fc3c2969c779d
使用conda的环境
snakemake -p --directory .test resources/mirna.fasta --use-conda --conda-frontend mamba -c5
结果显示如下
Building DAG of jobs...
Creating conda environment ../workflow/envs/env.yaml...
Downloading and installing remote packages.
在第一次使用conda环境的时候,会首先安装,该过程由于国内网速的原因,比较耗时间。这里我们可以手动创建conda的环境,具体的命令如下:
mamba env create --file workflow/envs/env.yaml --prefix .snakemake/conda/369d9fb0f93ac536da2fc3c2969c779d
注意
--prefix后面的内容是--list-conda-envs中查找到的,并且workflow/envs/env.yaml文件名称及内容一旦修改,369d9fb0f93ac536da2fc3c2969c779d这个hash值就会更改
如果你此时运行snakemake命令,仍旧会创建环境,此时需要运行下面命令创建一个空的文件夹
touch .test/.snakemake/conda/369d9fb0f93ac536da2fc3c2969c779d/env_setup_end
数据库下载
miRNA数据下载
miRNA的数据库地址: miRBase
可以使用下面命令下载
snakemake -p --directory .test resources/mirna.fasta --use-conda --conda-frontend mamba -c5
此时会得到三个文件
.
├── mirna.fasta
├── mirna_hairpin.fasta
└── mirna_mature.fasta
# mirna_hairpin.fasta
>hsa-let-7a-1 MI0000060 Homo sapiens let-7a-1 stem-loop
TGGGATGAGGTAGTAGGTTGTATAGTTTTAGGGTCACACCCACCACTGGGAGATAACTAT
ACAATCTACTGTCTTTCCTA
>hsa-let-7a-2 MI0000061 Homo sapiens let-7a-2 stem-loop
AGGTTGAGGTAGTAGGTTGTATAGTTTAGAATTACATCAAGGGAGATAACTGTACAGCCT
CCTAGCTTTCCT
>hsa-let-7a-3 MI0000062 Homo sapiens let-7a-3 stem-loop
GGGTGAGGTAGTAGGTTGTATAGTTTGGGGCTCTGCCCTGCTATGGGATAACTATACAAT
CTACTGTCTTTCCT
>hsa-let-7b MI0000063 Homo sapiens let-7b stem-loop
# head mirna.fasta
>hsa-let-7a-5p MIMAT0000062 Homo sapiens let-7a-5p
TGAGGTAGTAGGTTGTATAGTT
>hsa-let-7a-3p MIMAT0004481 Homo sapiens let-7a-3p
CTATACAATCTACTGTCTTTC
>hsa-let-7a-2-3p MIMAT0010195 Homo sapiens let-7a-2-3p
CTGTACAGCCTCCTAGCTTTCC
>hsa-let-7b-5p MIMAT0000063 Homo sapiens let-7b-5p
TGAGGTAGTAGGTTGTGTGGTT
>hsa-let-7b-3p MIMAT0004482 Homo sapiens let-7b-3p
CTATACAACCTACTGCCTTCCC
# mirna_mature.fasta 是上面两个文件合并后的文件
miRBase Sequence
hairpin.fa: Fasta format sequences of all miRNA hairpins(发夹结构)
mature.fa: Fasta format sequences of all mature(成熟) miRNA sequences
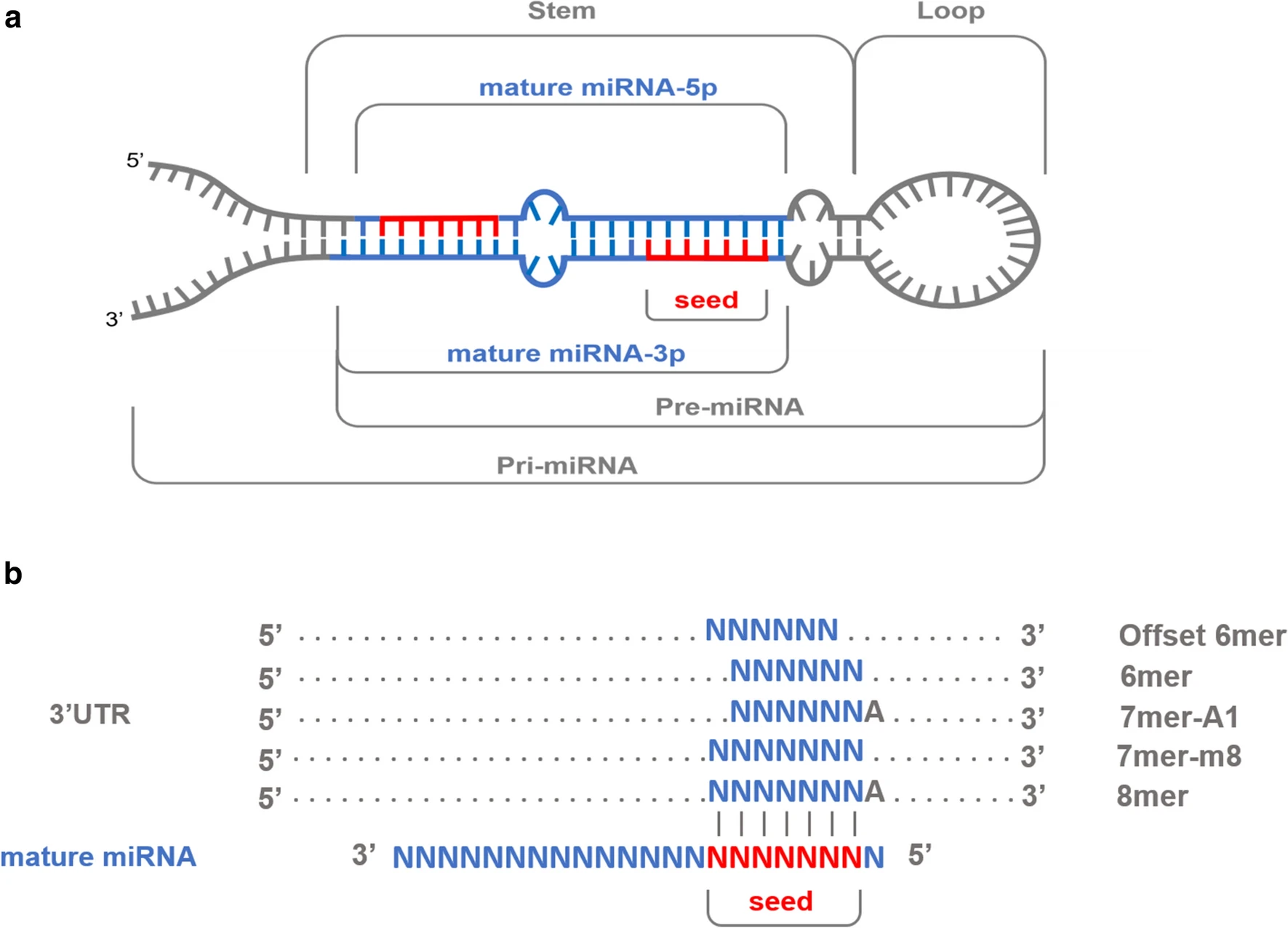
图a: 前体(precursor)miRNA序列发夹结构(hairpin conformation)的示意图,成熟的miRNA位于茎(蓝色突出显示)及其相应的种子区域(红色)[1]
图b: 典型的miRNA–mRNA相互作用,其中每个结合位点通过6至7个Watson–Crick配对(垂直线)与成熟miRNA的种子区(第2至第8个5′核苷酸)配对
piRNA数据下载
piRNA数据下载地址[2]: http://bigdata.ibp.ac.cn/piRBase/download/v3.0/fasta/hsa.v3.0.fa.gz
可以使用下面命令下载
snakemake -p --directory .test resources/pirna.fasta --use-conda --conda-frontend mamba -c5
此时会得到一个fasta文件
.
└── pirna.fasta
# head pirna.fasta
>piR-hsa-1
AGAAGACATTCGTGGAGGCGTC
>piR-hsa-2
ACGCCTCCACGTAGTGTCTT
>piR-hsa-3
ACGCCTCCACGAGTGTCTT
>piR-hsa-4
ACCAAGGACTTCATCTTCTCAGAGCTGCTGT
>piR-hsa-5
TCATCTTCTCAGAGCTGCTGTCCAACCTGC
建立index
miRNA建立index
snakemake --directory .test resources/mirna.1.bt2 --use-conda --conda-frontend mamba -c5 -p
得到以下文件
.
├── logs
├── mirna.1.bt2
├── mirna.2.bt2
├── mirna.3.bt2
├── mirna.4.bt2
├── mirna.rev.1.bt2
└── mirna.rev.2.bt2
piRNA建立index
使用下面命令建立piRNA的index
snakemake --directory .test resources/pirna.1.bt2 --use-conda --conda-frontend mamba -c5 -p
得到以下文件
.
├── pirna.1.bt2
├── pirna.2.bt2
├── pirna.3.bt2
├── pirna.4.bt2
├── pirna.rev.1.bt2
└── pirna.rev.2.bt2
下机fastq数据质控
merge_fastq_files
合并单个样本所有的fastq文件到一个文件中
snakemake --directory .test results/sampleA/fastq/sampleA_untrimmed.fastq.gz --use-conda --conda-frontend mamba -c5 -p
合并所有sampleA的fast1文件得到文件sampleA_untrimmed.fastq.gz
# zcat sampleA_untrimmed.fastq.gz | head
@Simulated1
CGAGGAACTGTAGGCACCATCAATAAAACCCTAGAC [AGATCGGAAGAGCACACGTCTGAAC] TCCAGTCACTTAGG
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEAEEEEEEEEEEEEEEAEEEEEEEAEEEEAEAEEAEEEEEEEEEEEE
@Simulated2
CAGTTTTGTCAGGTAAACTGTAGGCACCATCAATCCCAGTCGCTTC [AGATCGGAAGAGCACACGTCTGAAC] TCCA
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEAEEEEEEEEAEE<EAAAEEEEEEEA/EEEEEEAEEEEEAE
@Simulated3
TGCTGCTTTATTGTTATTGCTGCTTGATTGTTAACTGTAGGCACCATCAATATGAAGACGAATAGATCGGAAGAG
上面括号中的序列是Illumina的通用接头序列
trim_illumina_adapters
从reads的3'端去除Illumina的通用接头序列(Adapter sequences)
通用接头序列是: AGATCGGAAGAGCACACGTCTGAAC
snakemake --directory .test results/sampleA/fastq/sampleA_illumina_trimmed.fastq.gz --use-conda --conda-frontend mamba -c5 -p
结果得到文件sampleA_illumina_trimmed.fastq.gz
# zcat sampleA_illumina_trimmed.fastq.gz | head
@Simulated1
CGAGGAACTGTAGGCACCATCAAT [AAAACCCTAGAC]
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEAEEEEEE
@Simulated2
CAGTTTTGTCAGGTAAACTGTAGGCACCATCAAT [CCCAGTCGCTTC]
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEEEAEEEEEEEEAE
@Simulated3
TGCTGCTTTATTGTTATTGCTGCTTGATTGTTAACTGTAGGCACCATCAAT [ATGAAGACGAAT]
上面括号中的序列为
UMI
trim_umi_sequences
从reads的3'端去除UMI(Unique Molecular Identifier)序列,并将其添加到reads的头部
其中UMI的pattern为: NNNNNNNNNNNN
snakemake --directory .test results/sampleA/fastq/sampleA_umi_trimmed.fastq.gz --use-conda --conda-frontend mamba -c5 -p
结果得到文件sampleA_umi_trimmed.fastq.gz
# zcat sampleA_umi_trimmed.fastq.gz | head
@Simulated1_AAAACCCTAGAC
CGAGGAACTGTAGGCACCATCAAT
+
AAAAAEEEEEEEEEEEEEEEEEEE
@Simulated2_CCCAGTCGCTTC
CAGTTTTGTCAGGTAAACTGTAGGCACCATCAAT
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEE
@Simulated3_ATGAAGACGAAT
TGCTGCTTTATTGTTATTGCTGCTTGATTGTTAACTGTAGGCACCATCAAT
关于UMI在3'端的的原因,可以看下图,在illumina测序中reads是从5'向3'端测的。UMI是在adapter之后的,因此去除adapter后紧跟着的12bp为UMI的序列
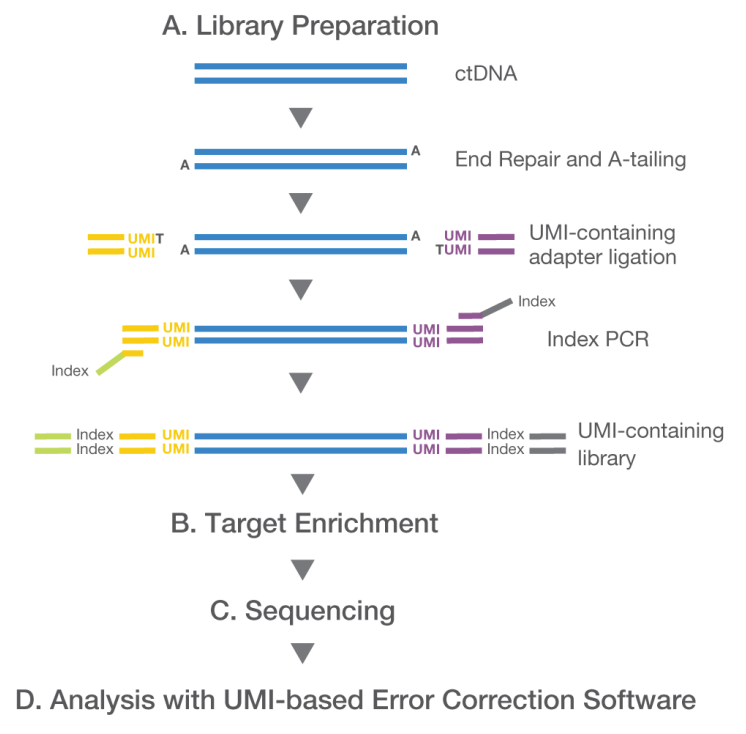
可以将UMI集成到Y-adapter连接中,而无需中断标准工作流程。在文库准备期间,原始文库准备套件的adapter简单地由UMI adadpter代替[3]。
trim_5_adapter
从reads 5'端去除接头
snakemake --directory .test results/sampleA/fastq/sampleA_5adapter_trimmed.fastq.gz --use-conda --conda-frontend mamba -c5 -p
结果得到文件sampleA_5adapter_trimmed.fastq.gz
# zcat sampleA_5adapter_trimmed.fastq.gz | head
@Simulated1_AAAACCCTAGAC
CGAGGAACTGTAGGCACCATCAAT
+
AAAAAEEEEEEEEEEEEEEEEEEE
@Simulated2_CCCAGTCGCTTC
CAGTTTTGTCAGGTAAACTGTAGGCACCATCAAT
+
AAAAAEEEEEEEEEEEEEEEEEEEEEEEEEEEEE
@Simulated3_ATGAAGACGAAT
TGCTGCTTTATTGTTATTGCTGCTTGATTGTTAACTGTAGGCACCATCAAT
trim_3_adapter
从reads 3'端去除接头,丢弃未修剪的reads并执行一些质量调整
snakemake --directory .test results/sampleA/fastq/sampleA_trimmed.fastq.gz --use-conda --conda-frontend mamba -c5 -p
结果得到文件sampleA_trimmed.fastq.gz
# zcat sampleA_trimmed.fastq.gz | head
@Simulated3_ATGAAGACGAAT
TGCTGCTTTATTGTTATTGCTGCTTGATTGTT
+
AAAAAEEEEEEEEEEEEAEEEEEEEEAEEAEE
@Simulated19_ATTCCCGCCGCG
GTTGATTCTGTGGGTGGTGGTGCAT
+
/AAAAAEEEEE/EEEEEAEEEEEEE
@Simulated31_TGACCCATCCCC
GATTTATGAAAGACGAACTACTGCCAAAG
比对reads到数据库
map_mirna_sequences
将reads比对到mirBase数据库序列,直接转换为排序的.bam文件,并生成.bai的索引文件。没有比对上的reads将保存为新的fastq 文件,随后与piRNA的数据库比对。
snakemake --directory .test results/sampleA/bam/sampleA_mirna.bam --use-conda --conda-frontend mamba -c5 -p
得到3个结果文件
sampleA_mirna.bai
sampleA_mirna.bam
sampleA_mirna_unmapped.fastq.gz
map_pirna_sequences
将reads比对到piRNAdb数据库序列,直接转换为排序的.bam文件,并生成.bai的索引文件。有比对上的reads将保存为新的fastq 文件,随后与参考基因组比对(本文没有执行该步骤)。
这里使用的序列是miRNA没有mapping上的文件:sampleA_mirna_unmapped.fastq.gz
snakemake --directory .test results/sampleA/bam/sampleA_pirna.bam --use-conda --conda-frontend mamba -c5 -p
得到3个结果文件
sampleA_pirna.bai
sampleA_pirna.bam
sampleA_pirna_unmapped.fastq.gz
合并bam文件
snakemake --directory .test results/sampleA/bam/sampleA_merged.bam --use-conda --conda-frontend mamba -c5 -p
得到两个文件
sampleA_merged.bai
sampleA_merged.bam
deduplicate_reads
使用UMItools dedup命令根据先前识别的UMI序列,去除重复的reads
snakemake --directory .test results/sampleA/bam/sampleA_merged_dedup.bam --use-conda --conda-frontend mamba -c5 -p
得到两个文件
sampleA_merged_dedup.bai
sampleA_merged_dedup.bam
获得count表
snakemake --directory .test results/sampleA/sampleA_counts_dedup.tsv --use-conda --conda-frontend mamba -c5 -p
得到文件sampleA_counts_dedup.tsv
# head sampleA_counts_dedup.tsv
Molecule Count
hsa-let-7f-5p 1
hsa-miR-16-5p 5
hsa-miR-21-5p 1
hsa-miR-24-3p 1
hsa-miR-28-3p 1
hsa-miR-92a-3p 1
hsa-miR-93-5p 3
hsa-miR-221-3p 1
hsa-miR-223-3p 1
运行所有样本
snakemake --directory .test --use-conda --conda-frontend mamba -c5 -p
最终得到两张表,其中counts.tsv没有运行deduplicate_reads和sampleA_counts_dedup两条命令。
# head counts.tsv
Molecule sampleA sampleB sampleC
hsa-let-7a-1 0.0 0.0 4.0
hsa-let-7a-2 4.0 4.0 0.0
hsa-let-7a-3 4.0 0.0 0.0
hsa-let-7b 12.0 4.0 4.0
hsa-let-7b-5p 0.0 4.0 0.0
hsa-let-7f-1 0.0 0.0 4.0
hsa-let-7f-5p 4.0 0.0 0.0
hsa-miR-103a-3p 0.0 0.0 4.0
hsa-miR-125a-5p 4.0 0.0 0.0
# counts_deduplicated.tsv
Molecule sampleA sampleB sampleC
hsa-let-7a-1 0.0 0.0 1.0
hsa-let-7a-2 1.0 1.0 0.0
hsa-let-7a-3 1.0 0.0 0.0
hsa-let-7b 3.0 1.0 1.0
hsa-let-7b-5p 0.0 1.0 0.0
hsa-let-7f-1 0.0 0.0 1.0
hsa-let-7f-5p 1.0 0.0 0.0
hsa-miR-103a-3p 0.0 0.0 1.0
hsa-miR-125a-5p 1.0 0.0 0.0















 695
695











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









