









论文解读:《DeepFormer: a hybrid network based on convolutional neural network and flow-attention mechanism for identifying the function of DNA sequences》
文章地址:https://academic.oup.com/bib/article-abstract/24/2/bbad095/7077271?login=false
DOI:https://doi.org/10.1093/bib/bbad095
期刊:BRIEFINGS IN BIOINFORMATICS
2022年影响因子/JCR分区:13.994/Q2
发布时间:2023年2月21日
Github: https://github.com/YZ20211221/DeepFormer
1.文章概述
准确识别DNA序列的功能是基因组领域一项重要且具有挑战性的任务。到目前为止,深度学习已广泛应用于DNA序列的功能分析,包括DeepSEA,DanQ,DeepATT和TBiNet。然而,这些方法存在计算复杂度高、未充分考虑染色质特征之间远距离相互作用等问题,从而影响预测精度。在这项工作中,作者提出了一种混合深度神经网络模型,称为DeepFormer,基于卷积神经网络(CNN)和 flow-attention mechanism 进行DNA序列功能预测。在DeepFormer中,CNN用于捕获DNA序列的局部特征以及重要的基序。基于flow网络守恒定律, flow-attention mechanism 可以捕获更多具有线性时间复杂度的序列特征之间的远端相互作用。作者使用近919万个非编码DNA序列4个染色质特征的常用数据集将DeepPreformer与上述四种经典方法进行了比较。实验结果表明,DeepFormer 明显优于四种方法,平均召回率至少比其他方法高 9.7%。此外,作者还证实了DeepForformer使用阿尔茨海默病,α-地中海贫血的致病突变和CCCTC结合因子(CTCF)活性修饰捕获功能变异的有效性,进一步预测了五种组织的玉米染色质可及性,并验证了DeepFormer的可推广性。DeepForformer 的平均召回率比经典方法高出至少 058.1%,显示出很强的鲁棒性。
2.关键点
- 基于CNN和流注意机制构建用于识别DNA序列函数的DeepFormer模型。 CNN 模块捕获 DNA 序列的局部特征(motifs),flow-attention 机制模块捕获长距离 DNA 序列特征之间更远端的相互作用。
- DeepFormer 在平均准确率和召回率方面优于现有的经典模型(DeepATT、DanQ、DanQ_JASPAR、DeepSEA 和 TBiNet)。
- 评估DeepFormer 挖掘影响疾病的重要基序和潜在突变位点的能力。
- 使用五种玉米组织的染色质可及性预测数据集验证了DeepFormer 的泛化能力。
3.背景
绝大多数人类基因组位于非编码区,大多数与疾病相关的常见遗传变异位于非编码区。许多研究报道,转录因子 (TF) 结合受多种因素的影响,例如染色质可及性程度、TF 结合位点的序列特异性和结合位点的可变性。 DNase I 超敏位点和组蛋白标记可能具有更复杂的基因组序列特异性潜在机制。也有报道称,基因组序列中存在可以表征序列特征(例如顺式调控元件)的模式,这有助于解析非编码序列的功能。近年来,研究人员利用机器学习的方法,对已知函数的基因组序列进行模型训练,然后对输入的DNA序列进行序列函数预测。为此,开发能够准确识别DNA序列功能的模型不仅对功能基因组研究有意义,而且对当前的生物学研究也很有价值。
DeepSEA和 DeepBind是最早将深度学习应用于基因组序列分析的代表性研究。这种方法首先使用onehot编码对基因组序列进行编码,然后通过建立深度学习模型来预测生物功能。最大的优点是只需要输入基因组序列,不需要输入其他特征数据,可以提取序列中的依赖性、复杂性和局部性特征。实验结果还表明,深度学习方法的准确率远高于其他机器学习方法。自 2015 年以来,研究人员将深度学习应用于人类生物学功能的分析,包括序列功能预测、甲基化状态、可变剪接、增强子序列、染色质可及性和 TF 结合位点。DanQ 可以通过结合 CNN 和 BiLSTM 来捕获序列中的基序和长距离依赖性以进行 DNA 序列功能预测。注意力机制从具有仿生概念的数据中过滤掉高价值信息,使不适合注意力模型的内容变弱或被遗忘。关于每个特征之间的相互关系,它可以捕获远距离特征之间的关系。 TBiNet 主要使用 CNN、自注意力机制和 BiLSTM 网络构建模型,成功地提高了 TF 结合位点的预测准确性。此外,DeepATT模型包含CNN、BiLSTM网络、类别注意力神经网络和类别密集神经网络。DeepATT 可以有效地识别调控功能并挖掘非编码 DNA 序列中不同特征之间的重要相关性。
self-attention机制模块在取得优异性能的同时,有效解决了BiLSTM的不足。但是self-attention机制的计算复杂度为O(n2),输入数据长度为n,导致在构建模型时消耗大量资源。此外,这些方法没有充分考虑染色质特征之间更远端的相互作用,从而影响预测准确性。为了进一步降低计算复杂度并提高预测精度,作者提出了一种基于 CNN 和流动注意力机制的用于 DNA 序列功能预测的混合 DNN 模型(DeepFormer)。
- DeepFormer 中的 CNN 模型用于捕获 DNA 序列的局部特征以及重要的序列基序。基于flow网络的守恒定律,作者充分利用竞争和分配机制来获得流动注意力的注意力图,以捕捉染色质特征之间的相互作用。该模型能够以线性时间复杂度(O(n))捕获长距离DNA序列特征之间更远的关系,在相同内存空间的条件下比self-attention机制和BiLSTM更有效。
- 在一个常用的 919 人类染色质特征预测数据集上的实验结果表明,DeepFormer 在平均准确率和召回率方面优于现有的经典模型(DeepATT、DanQ、DanQ_JASPAR、DeepSEA 和 TBiNet)。此外,作者使用染色质可及性预测的玉米五个组织数据集验证了 DeepFormer 的泛化能力。
- 使用 DeepFormer 的可视化方法对挖掘一些重要的基序进行分析,并对非编码 DNA 功能效应进行解释。阿尔茨海默病、α-地中海贫血致病突变和 CCCTC 结合因子 (CTCF) 活性变化用于验证关于 DeepFormer 的 DNA 序列功能变异的可解释性。
4.数据
作者使用与 DeepSEA、DanQ 和 DeepATT 相同的数据集来分析和比较 DeepFormer 的性能。数据集是从 DeepSEA 网站下载的。简而言之,人类 GRCh37 参考基因组被分成不重叠的 200 bp 区间。通过取 ENCODE 和 Roadmap Epigenomics 的 ChIP-seq 和 DNaseseq 的 919 个峰值的交集,为每个样本生成一个长度为 919 的二进制目标向量。919 个染色质特征由三类组成:(1) 125 个 DNase I 敏感性特征,(2) 690 个 TF 结合特征和 (3) 104 个组蛋白标记谱特征。每个样本的输入是一个 1000 bp 的序列,以 200 bp 的 bins 为中心,该 bins 与至少一个 TF 结合 ChIPseq 峰重叠。存在于其 200 bp 的 bins 中一半的峰区域标记为 1;否则,它被标记为 0。
数据集分为不重叠的训练集、验证集和测试集。作者选取7号染色体上的 8000 个样本作为验证集,选取8号和9号染色体上的 45524 个样本作为测试集。选择剩余染色体上的 4400000 个样本作为训练集。在构建数据集时,同时考虑了 DNA 的正序列和反向互补序列。因此,实验的数据集数量多于ENCODE 和 Roadmap Epigenomics 中的数据集。 One-hot 的 encoding后,每个1000 bp 的DNA序列用一个1000×4的二元矩阵表示,四列分别对应A/G/C/T。
5.方法
DeepFormer 由四部分组成:输入层、CNN 层、flow-注意力层和输出层。 CNN层主要用于实现序列中的特征提取和捕获图案。作为注意力机制的改进,flow-注意力层基于网络流动守恒原理重新整合信息流,该方法有利于保证整个算法消耗线性复杂度的计算时间,增强提取DNA序列重要远端特征的能力。 DeepFormer的流程图如图1所示,模型的输入是长度为1000 bp的DNA基因组序列。 A编码为[1,0,0,0],T编码为[0,1,0,0],C编码为[0,0,1,0],G编码为[0,0 ,0,1]。 one-hot编码后得到一个1000*4的on e-hot编码矩阵作为CNN层的输入。

输出层使用两个完全连接的层对 919 个染色质特征进行分类。使用 sigmoid 获得 919 个染色质特征(125 个 DNase I 敏感性特征,690 个 TF 结合特征和 104 个组蛋白标记谱特征)的概率,其范围在 0 和 1 之间
6.结果
6.1 消融实验

6.2 AUROC 和 AUPR 结果的比较

绘制了每个染色质特征的 AUROC 和 AUPR 结果,如图 2C 和 D 所示。总体而言,DeepFormer 的 919 个染色质特征预测结果更集中在高值区域。
6.3 DeepFormer的ROC曲线和PR曲线的结果
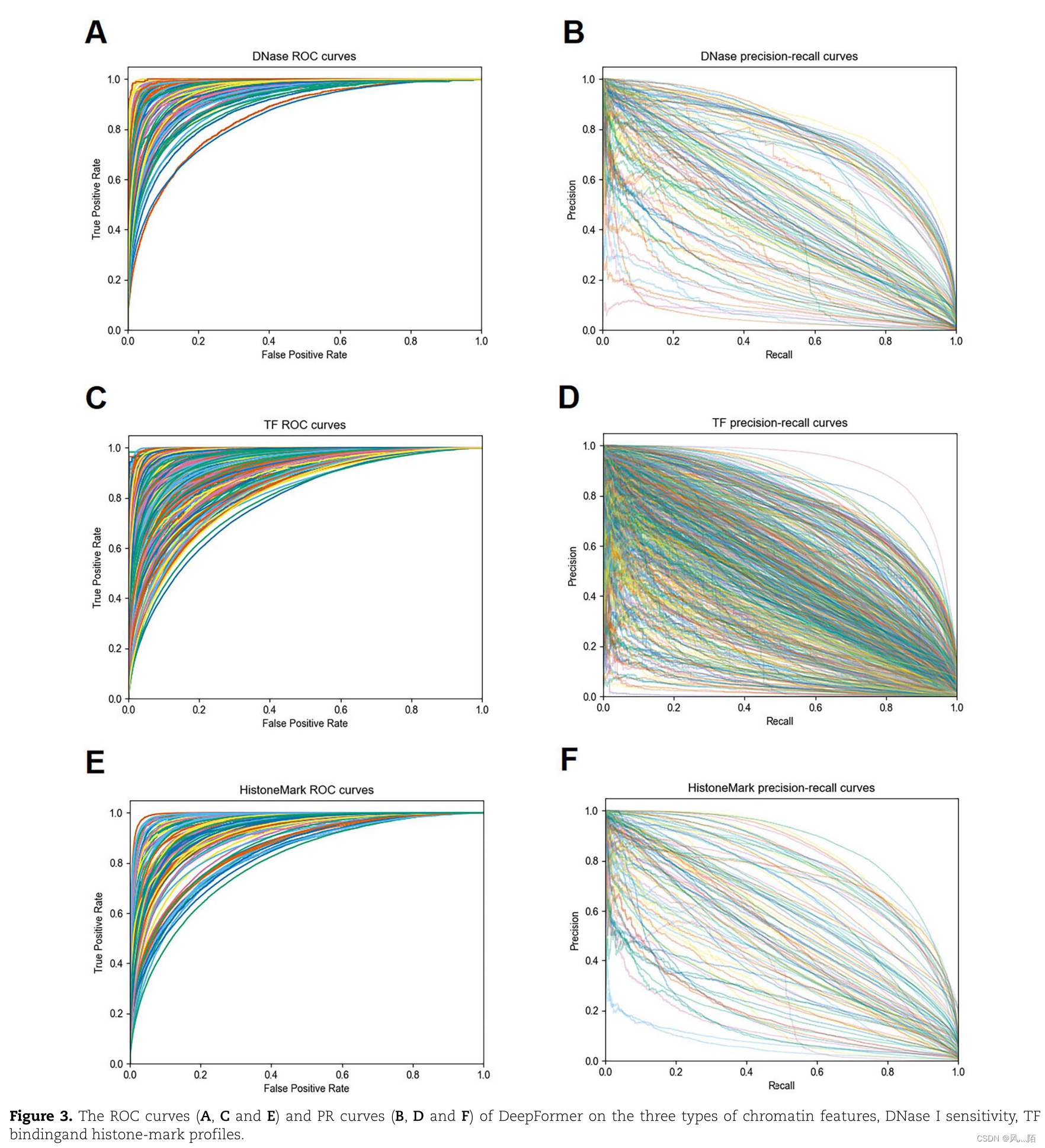
作者绘制了 DeepFormer 在预测 DNase I 敏感性、TF 结合和组蛋白标记配置文件中的 ROC 和精确率与召回率 (PR) 曲线。
6.4 DeepFormer 与 DeepATT 的比较

作者比较了 DeepFormer 和 DeepATT 的 AUROC 和 AUPR。对于大多数染色质特征,可以看出 DeepFormer 明显优于 DeepATT。
6.5 Motif分析
将内核从 DeepFormer 的卷积层转换为序列中特定位置的位置特定权重矩阵(PSWM)或图案。作者使用 TOMTOM 比较算法来搜索这些潜在图案与已知图案的匹配项 (JASPAR 2020 [27]),并通过计算图案之间的 Pearson 相关系数来评估相似性。结果表明,在 DeepFormer 学习到的 320 个 motif 中,有许多重要的潜在 motif 可以匹配已知的 motif (E < 0.01)。DeepFormer中的卷积层可以捕获非编码DNA序列函数在不同位置的特异性信息。

6.6 变异效应评价
作者使用三个实际案例来说明 DeepFormer 在发现影响疾病的突变位点方面的有效性。
(1)图 6A 描述了 DeepFormer 模型的 DNA 序列饱和突变结果。在染色体 16:20970 中的碱基 T 到碱基 C 的突变中发现了最高的效应值,这表明 DeepFormer 准确预测了α地中海贫血的致病突变。
(2)HepG2细胞系特征性CEBPB的TTGACTCAA序列109817590染色体1中碱基G突变为T。 DeepFormer 模型在 DNA 序列的饱和突变结果中获得了更高的效应值。
(3)染色体X:73072592中碱基G突变为碱基C的增益得分最高,增强了CTCF的结合能力。

6.7 不同方法的特征捕获和复杂度分析
919个染色质特征的多任务预测模型的主要优点是它可以找到919个染色质特征之间的潜在关系以及DNA序列特征之间更远的关系。


为了进一步证明捕获不同方法的 919 个染色质特征之间关系的能力,作者将 DeepSEA、DanQ、TBiNet、DeepATT 和 DeepFormer 的 919 个染色质特征的成对 Spearman 相关性的秩变换可视化,如图 8 所示。在图 8 中,可以看出不同的方法可以有效地捕获染色质特征之间的关系。

计算了不同方法预测结果矩阵与实验结果矩阵的Spearman相关性和余弦相似性的均值和中值。表 3 和表 4 显示了关于四组染色质特征(919 个染色质特征、690 个 TF 结合特征、125 个 DNase I 敏感性特征和 104 个组蛋白标记谱特征)的详细结果。

6.8 模型泛化能力评估

7.总结
作者提出了一种混合 DNN 模型 (DeepFormer),用于基于 CNN 和流动注意力机制的 DNA 序列功能预测。 CNN 可以捕获 DNA 序列的局部特征,flow注意机制可以捕获输入 DNA 序列中更远端特征之间的关系,使模型更加关注具有重要调节功能的基序位置。在常用的919个染色质特征数据集上的实验结果表明,DeepFormer的准确率优于DeepSEA、DanQ、DeepATT和TBiNet。作者使用染色质可及性预测的玉米五个组织数据集来验证 DeepFormer 的泛化能力。此外,作者还使用三个案例来解释挖掘重要基序以解释非编码 DNA 功能效应的能力。

















 1778
1778

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









