







下面是我对GLM模型的理解:
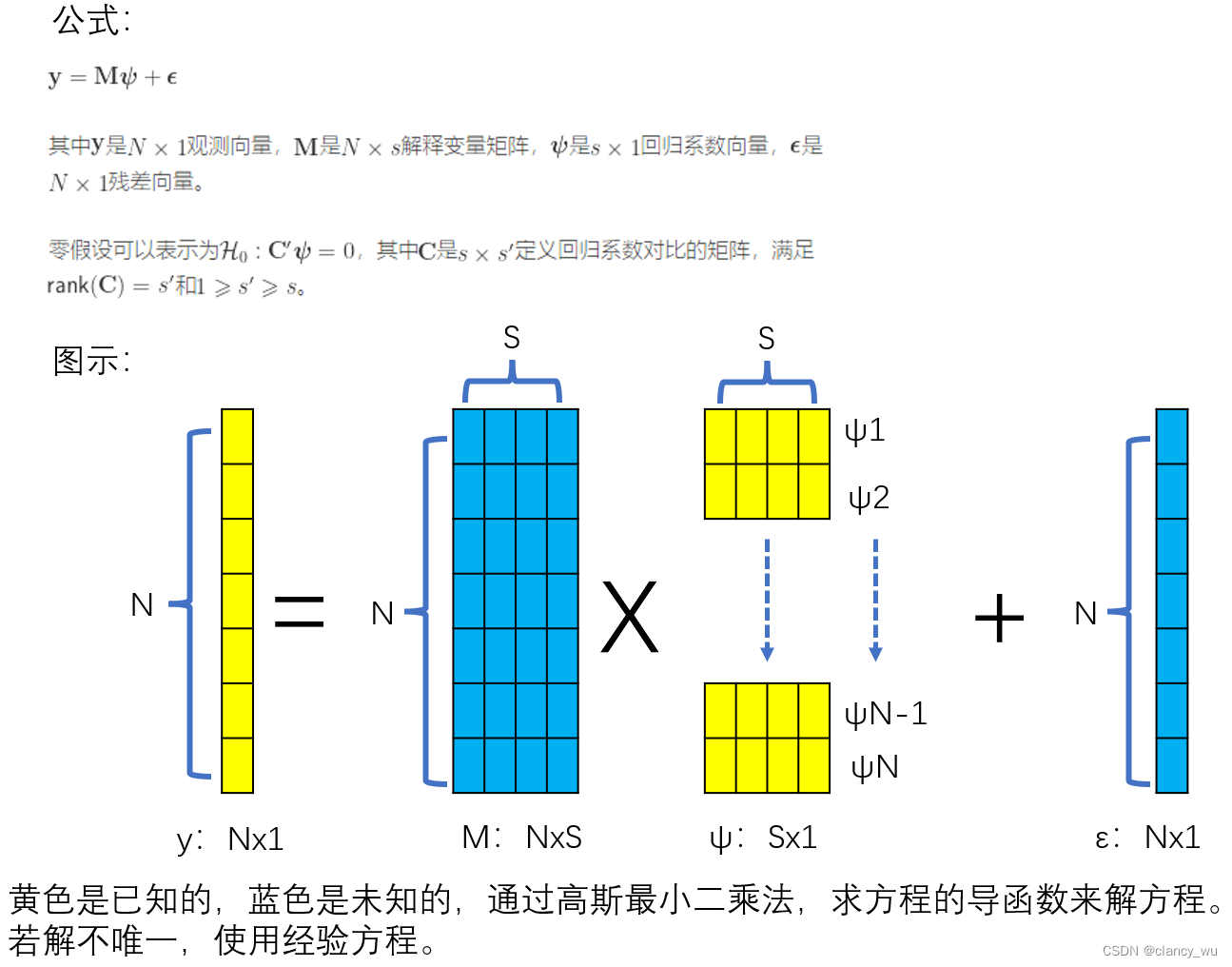
数据编码的方式
在一般统计中,常用的coding方式有dummy,effect和cell.mean,这个在R和python中都可以实现。
dummy coding 举例
假设有4个组别A, B, C, D,它的自由度是4-1=3,因此它可以用3个不同位置的1来编码代表4个组(有一个组作为reference组,其编码全为0).
假设如下的表格数据:

把g4组作为参考组,使用dummy coding转换后如下:
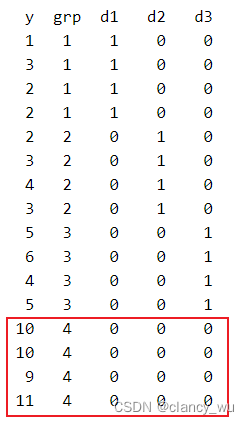
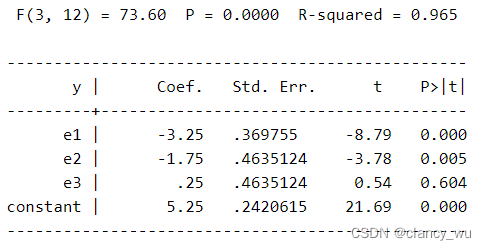
如此以后,进行F检验,得到的值,其截距就是参考组的平均值。而其他系数就是其均值差,例如-2=8-10,-7=3-10等等。
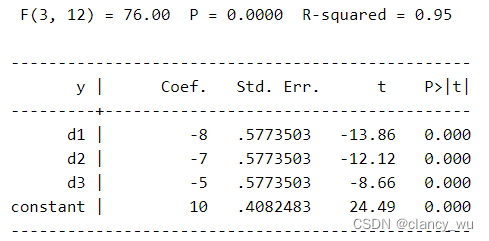
这个在python中就是独热编码过程。
R中就是如下:
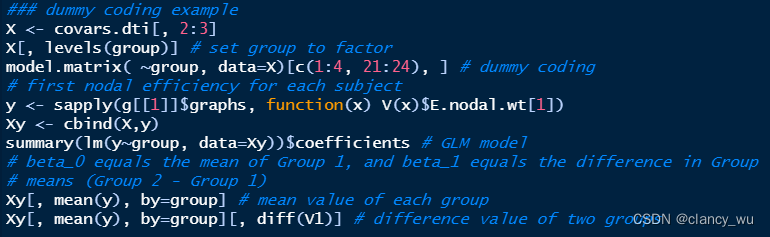
effect coding 举例
还是考虑上述数据,使用effect coding转换后如下:
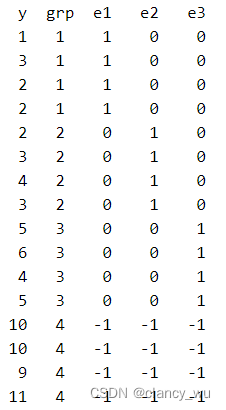
同样以g4组为参考组,所以能够看出,effect coding 和 dummy coding的区别在于,前者参考组为[-1, -1, -1]的矩阵,后者参考组为[0, 0, 0]的矩阵,其他就没有区别。

在effect coding后,算出来的截距是所有组平均值的平均值,即
y
‾
\overline{y}
y (等于5)。这个时候每组的coef,是其组的平均值和所有值的总平均值(constant值)的差值,即-3=2-5, -2=3-5。
不平衡的数据使用effect coding的注意事项
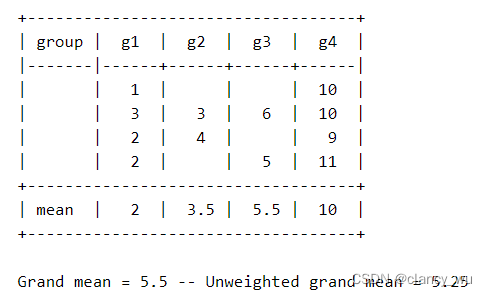
不平衡数据算出来的截距,是所有组平均值的平均值,而不是所有观察值的平均值。在这个数据中,一共有12个观察值,把12个值相加除以12,其平均值是5.5。而若把每一组进行组内平均,再把所有组的平均值进行平均,得到的值就是5.25,也就是F检验中的截距。
这个是需要注意和记住的地方。通过这个例子,也会对参数检验中对比均值的F检验有一个更为深刻的认识,即不管数据平衡与否,它只比较每组的平均值有无差异。很好用。
如何选择dummy coding和effect coding,两者区别?
如果模型中有多个分类变量,dummy coding 和 effect coding没有区别,只需要注意截距和系数的意义就行。
但是如果只有2种分类变量,dummy coding (编码0)和effect coding(编码-1)是有区别的,即effect coding的编码方式可以分析两个变量的交互效应。设想一下,当分类变量只有2个时,比如病人组和健康人组,那么使用dummy coding只有1和0,而使用effect coding 有1和-1,中间还有0态,所以effect coding可以分析交互作用,这样去记忆。因此这个时候使用effect coding可以获得对简单效应和交互效应的合理估计。
仔细回想一下,FSL的FEAT模块就是通过上述的方式去编码matrix design的。
R中如下:
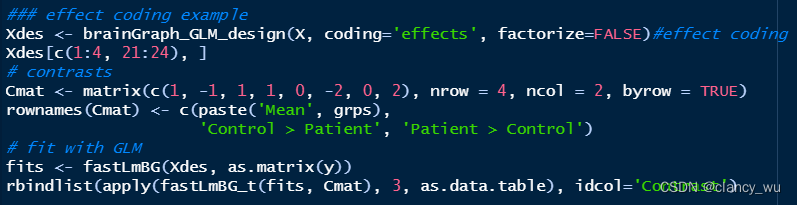
fastLmBG (fast lm brain graph) solve the least squares problem to estimate model coefficients, residuals, etc. for brain network data.
fastLmBG_t and fastLmBG_f calculate contrast-based statistics for T or F contrasts, respectively. It accepts any number of contrasts (i.e., a multi-row contrast matrix).
cell means coding 举例
SPM里的2-nd-level comparison就是cell means coding方法,这个方法只用于组别为2的数据。
因为只有2个组,所以不需要设置参考组,所以就是用[1, 0]和[0, 1]来表示两个组,然后design matrix对应着就是1-0=1和0-1=-1。
与上述两个方法相比,这个是对比均值差异,没有设置参考组,较为简单。
- 单元格均值模型仅用于使估计语句中的对比更易于编写的目的。唯一考虑的输出部分是与对比度估计相关的部分(这通常位于输出的底部)。
- 用单元格编写估计语句意味着模型更容易,因为它只包含一个向量(用于最高阶交互)。使用分析模型,同一对比度的估计语句可能包含多个向量和/或矩阵,因此更难以正确指定。
在R中的实现:
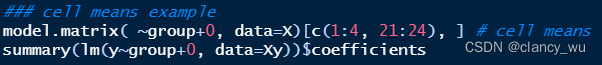
model.matrix是R自带的base函数,可以实现dummy和cell means coding,前者就是常规的 y —— g r o u p y —— group y——group,后者是 y —— g r o u p + 0 y —— group+0 y——group+0。
brainGraph_GLM_design是brainGraph包的函数,其中coding方式可以选择dummy,cell meaning 和 effects三种方式。
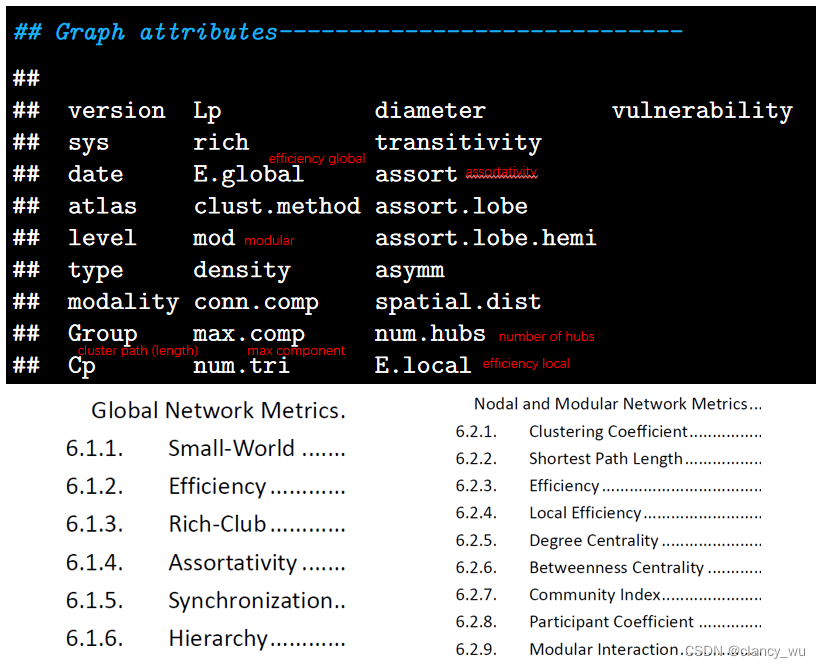
图论中的常用指标
图的常用指标
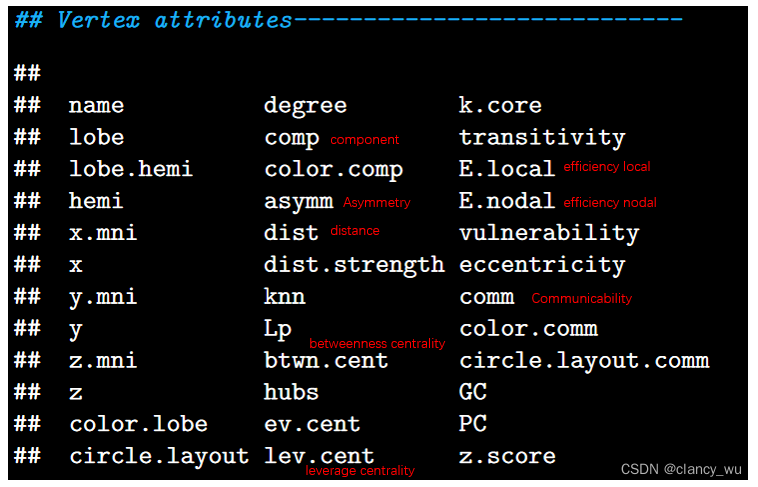
顶点的常用指标
边的常用指标
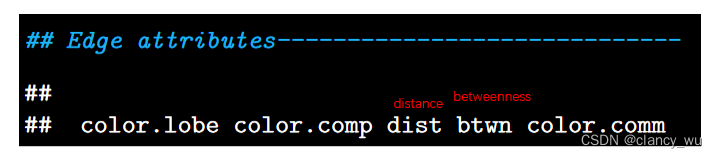
以上这些指标还可以加入.wt来查看weighted值和非weighted值(binary)。
GLM实例
我们在创建图的时候,常规会使用50个阈值【0.10,0.11.。。0.49,0.50】去生成图,DPABI和GRETNA设置的阈值参数会有些许差异,但是过程都是一样。
在GLM分析中,我们只能在单一阈值下去使用GLM模型去比对单一指标。在使用GLM比较时,一共有2步需要做。第一是创建design matrix,第二是根据design matrix进行GLM统计分析。(整个过程跟FSL的FEAT一模一样,可以相互参考)
以我现在手头的数据为例,我的数据是34:37的数量,healthy group和patient group。
两组比较-不考虑混淆因素
(1)首先是基础组别信息
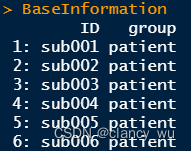
(2)根据组别信息创建design matrix
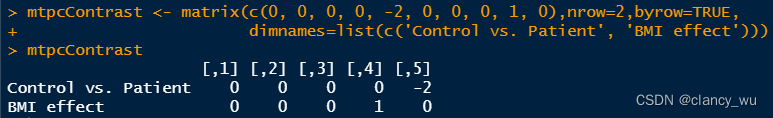
CompareMatrix <- matrix(c(0, -2), nrow = 1, dimnames = list('Control > Patient'))
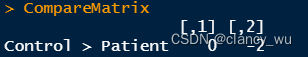
(3)根据design matrix,进行GLM分析
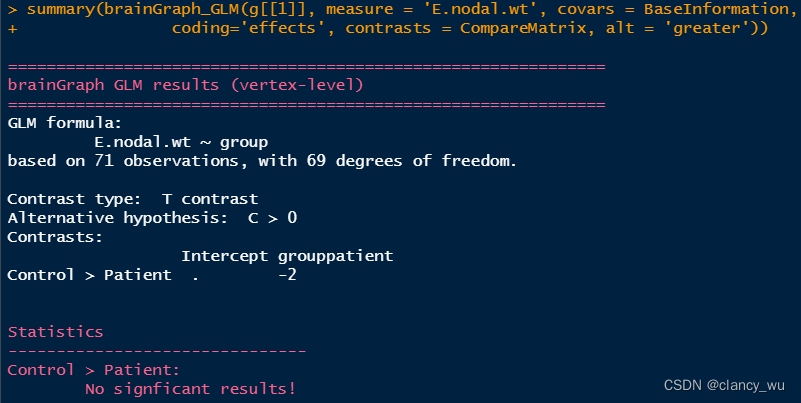
上面是单侧的分析。其实双侧的分析与上面同理,比较简单,双侧分析如下:
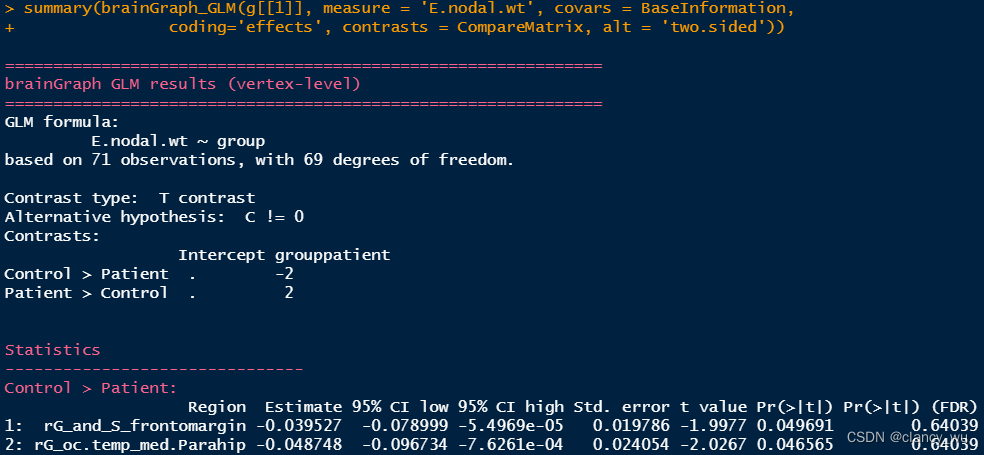
比较好的一点是,它会告诉FDR校正的结果。
两组比较-考虑混淆因素
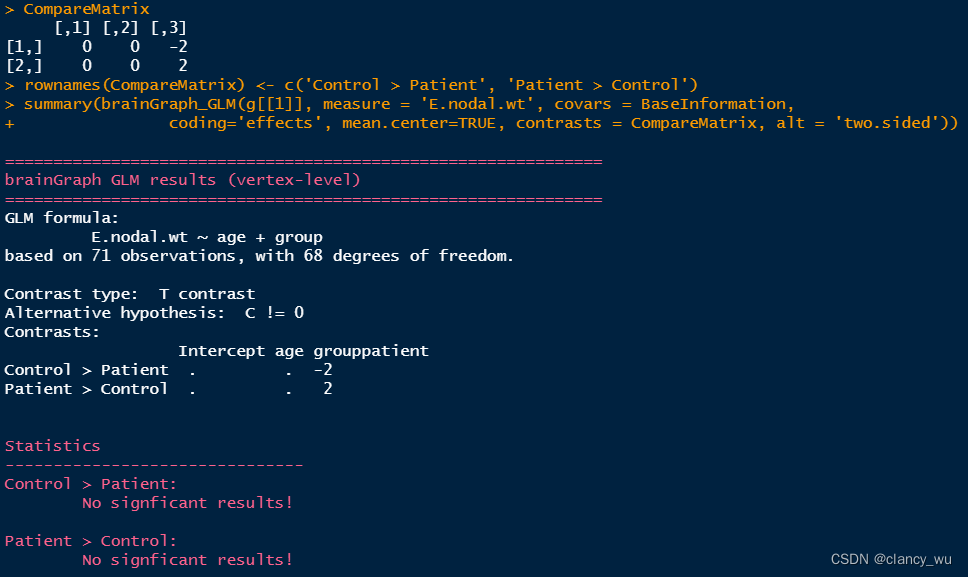
值得注意的是,有多少变量,CompareMatrix就要有多少列,函数会自动把group放到最后作为对比。
两组比较-考虑交互效应
如果要考虑交互效应,那么在design matrix时就与上述描述有点区别
计算group与连续变量交互的效应
连续变量大家应该清楚,分类变量是变量没有大小意义,连续变量是变量呈现连续表现,有大小意义。
以年龄举例,假如要做年龄和组别的交互效应:
(1)Base Information
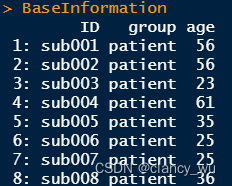
(2)design matrix
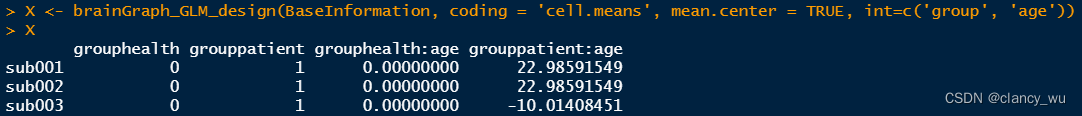
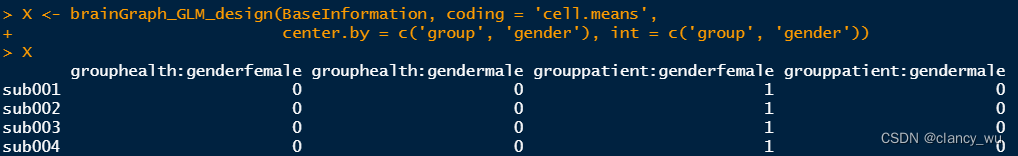
这里用的cellmeans编码方法,所以分为了两组,如果是effect编码,就是一组变量。
数据的标准化有好几种,常用的有:中心化,标准化和归一化。
- 归一化: x = ( x − m i n ) / ( m a x − m i n ) x = (x - min) / (max - min) x=(x−min)/(max−min)
- 标准化: x = ( x − m e a n ) / s t a n d a r d d e v i a t i o n x = (x - mean) / standard deviation x=(x−mean)/standarddeviation
- 中心化:
x
=
x
−
m
e
a
n
x = x - mean
x=x−mean
R语言中自带scale函数,中心化使用scale (x, center=T, scale=F),标准化使用scale (x, center=T, scale=T),归一化没有现成的函数,因为太简单了,自己编写+lappy批量处理就行。
这里的mean…center就是中心化处理,并且是cross group。如果要限定组内,就要center.how=‘within-group’,但是一般不建议这样处理,除非你有特殊需求。因为介绍到这里了,所以把附带的知识点也提了下。
然后int就是interaction,交互的变量,这里我做的就是group 和 age。
(3)compare matrix
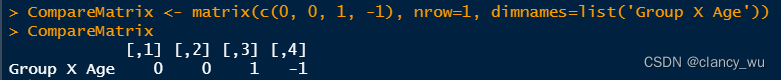
(4)GLM拟合
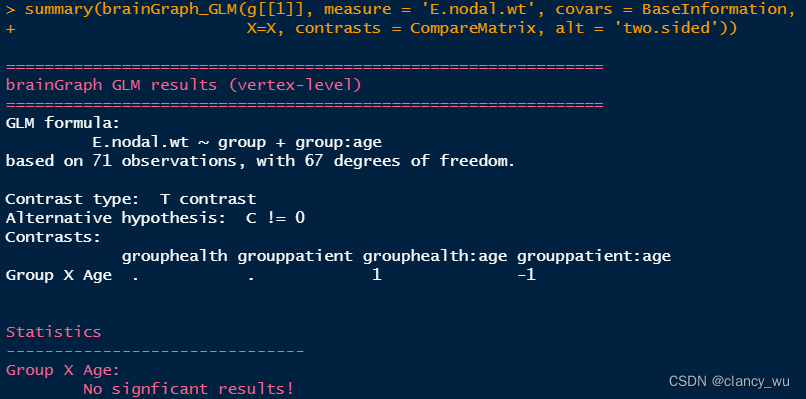
计算group与分类变量交互的效应
(1)Base Information
(2)Design Matrix
(3)compare matrix
(4)GLM
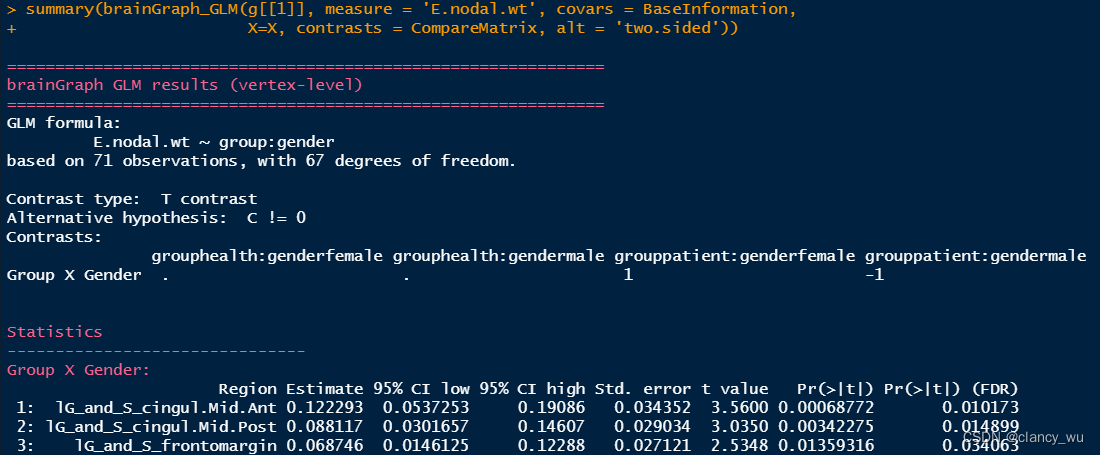
GLM中的置换检验实例
GLM的实现底层还是T检验(默认是T检验,所以结果中有T值,也可以自己选择F检验),这种其实还是有一种正态性假设在里面,属于参数检验。与GLM对应的,有一种不假设的非参数方法,就是GLM结合置换检验。
上述使用的GLM模型属于传统的GLM,即使用最小二乘法去求解方差系数(求导)。还有一种方法就是GLM模型结合置换检验,即使用置换检验法去求解方程系数。这种有3个方式:Freedman-Lane (the default), Smith, and Ter Braak。具体区别见(doi:10.1016/j.neuroimage.2014.01.060.),中文也有介绍:

(1)选择需要纳入分析的混淆变量
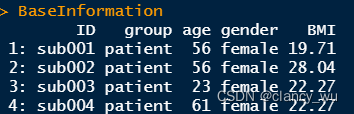
(2)进行编码
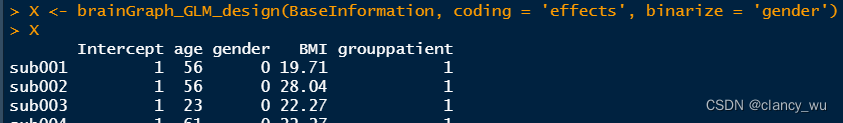
这里如果有多个混淆的分类变量,可以binarize = c(‘X’, ‘X’)。
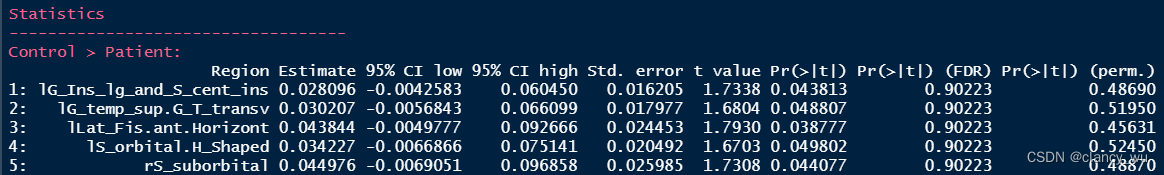
(3)对比的方式

health group是-1,patient group是1,所以control > patient 就是 -2。
(4)GLM分析
(5)可视化
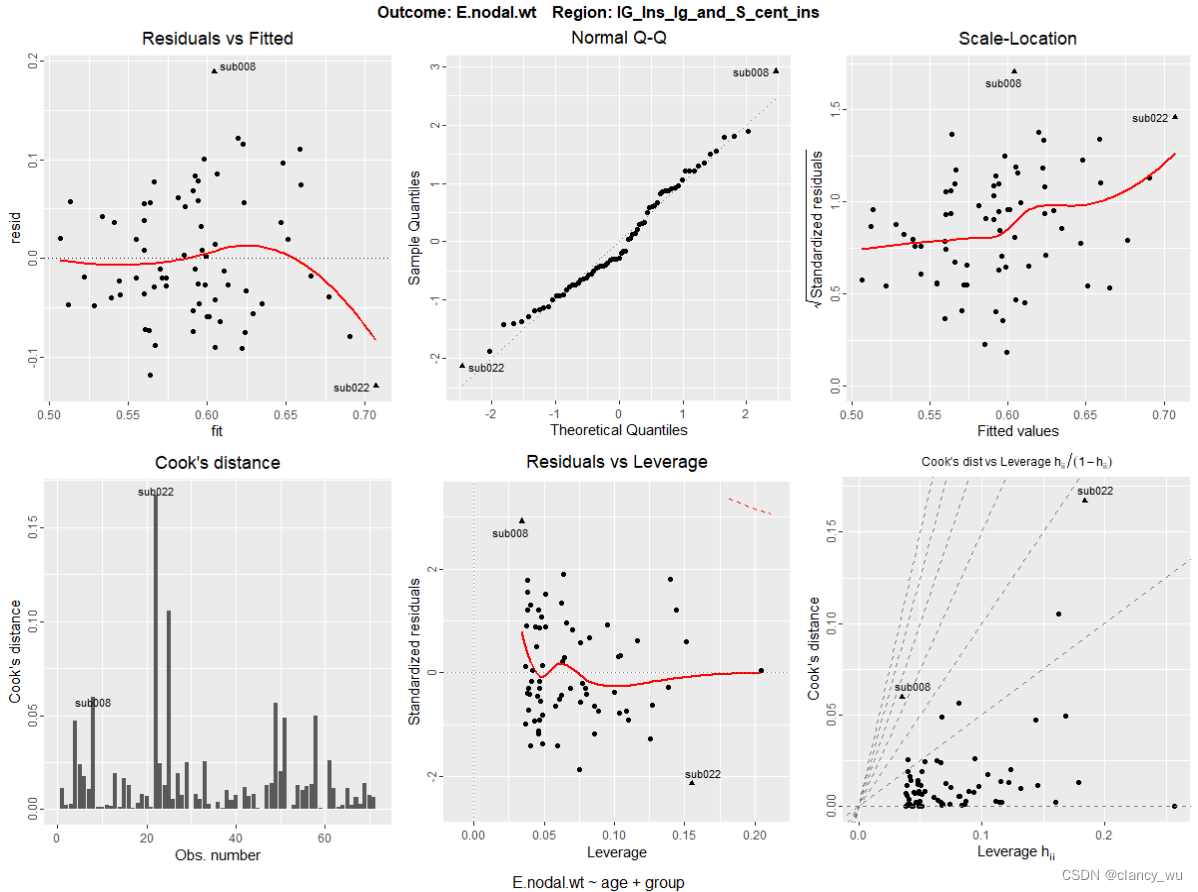
下面是单个ROI:
grid.arrange(plot(diffs.perm, region='lG_Ins_lg_and_S_cent_ins', which=1:6)[[1]])
如果是全部ROI都画,就:
glmPlots <- plot(diffs.perm, which=1:6)
ml <- marrangeGrob(glmPlots, nrow=1, ncol=1)
ggsave('glmPlots.pdf', ml, width=8.5, height=11)
glmPlots会循环的画出所有ROI。
multi-threshold permutation correction MTPC 多阈值置换校正
2015年Mark Drakesmith提出了这种 multi-threshold permutation correction MTPC的方法(doi:10.1016/j.neuroimage.2015.05.011)来克服图论方法的假阳性率。
- 误报 (FP) 对图论 (GT) 分析具有深远影响。
- 阈值可以减少 FP 的影响,但会引入其自身的偏差。
- GT 指标的统计数据在阈值之间变化很大。
- 使用新的排列方法 (MTPC) 可以可靠地检测群体效应。
- MTPC 比现有的曲线下面积 (AUC) 方法更灵敏和稳健。
由于在计算脑连接的时候,有一些虚假连接(如纤维束中的虚假追踪和功能连接中的虚假连接),会导致一些不符合实际的过于高估的密度(density),所以需要用阈值网络。网络的连接方式有FA均值、纤维束流线、FC等,都不相同,所以没有一个准则去决定使用哪一种阈值序列。但是使用了阈值网络和MTPC整体而言结果的假阳性会小很多。所谓的阈值,就是说在该阈值上的连接被丢弃,只计算该阈值下的网络属性。
MTPC的使用过程:
- 定义一个阈值序列,在每个阈值下计算所有被试的网络属性
- 对所有阈值下的所有被试网络进行MTPC统计,配合GLM模型
- 在每一个阈值下进行置换检验,比较组群差异 S o b s S_{obs} Sobs
- 在每个阈值下的每次置换检验中,使用最大统计数的零分布统计值(null distribution)作为是否显著的衡量标准
- 根据null distribution值的衡量结果,取前百分之α的统计值,作为critical value, S c r i t S_{crit} Scrit
- 比较cluster中, S o b s S_{obs} Sobs 和 S c r i t S_{crit} Scrit 的大小,并且计算AUC值(这个AUC其实就是MTPC的AUC值)
- 计算critical AUC值 A c r i t A_{crit} Acrit
- 比较
A
c
r
i
t
A_{crit}
Acrit 和
A
M
T
P
C
A_{MTPC}
AMTPC的大小,然后决定是否接受零假设。
边指标计算和节点指标计算都是一样的流程,只不过节点计算时,需要更大的多重校正。
多指标MTPC计算的实例:
(1)创建要分析的表格。
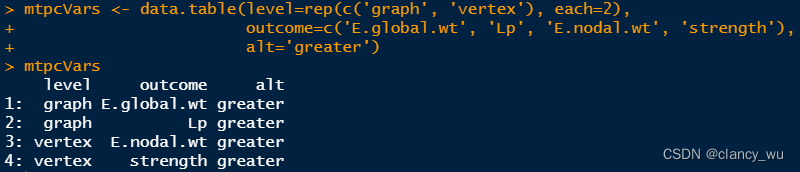
(1-2)假如要修改数据框,可以使用data.table的函数,即:=代表赋值。我不太熟悉这个,我用tidyverse比较多一些。
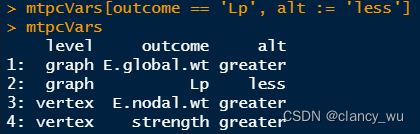
(2)为每一个设置permutation次数。
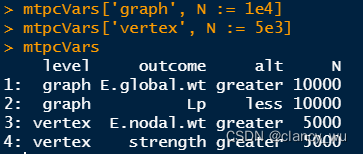
(3)设置置换矩阵
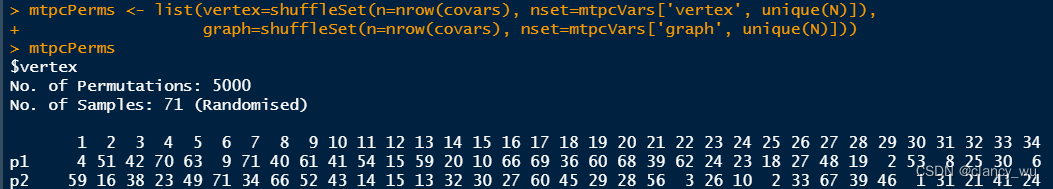
nrow(covars)就是有多少行数据,一般都返回的是受试者数量。然后nset是生成了1-N的顺序矩阵整数数字,然后再用shuffleSet函数把矩阵打乱。这样就得到了所有被试的置换矩阵。
(4)生成设计矩阵
(5)循环的在每一个被试水平进行指标的计算
mtpc.diffs.list <- sapply(mtpcVars[, unique(level)], function(x) NULL)
for (x in names(mtpc.diffs.list)) { # Loop across 'level'
# The 2nd-level is for each network metric (for the given level 'x')
mtpc.diffs.list[[x]] <- sapply(mtpcVars[x, unique(outcome)], function(x) NULL)
for (y in mtpcVars[x, outcome]) {
# Print some timing info in the terminal; optional
print(paste('Level:', x, '; Outcome:', y, ';', format(Sys.time(), '%H:%M:%S')))
mtpc.diffs.list[[x]][[y]] <-
mtpc(g, thresholds, covars=covars.dti, measure=y, contrasts=mtpcContrast,
con.type='t', level=x, N=mtpcVars[.(x, y), N], perms=mtpcPerms[[x]],
alt=mtpcVars[.(x, y), alt])
}
}
- 创建list,里面为NULL,作为后面储存数据的容器
- names(mtpc.diffs.list)是vertex和graph,即在level水平进行循环
- outcome是具体指标,从y开始就是循环进行指标的计算
- mtpc就是函数主体,其参数和用法与GLM相同,很简单。
运行起来还算挺快的,10000次的置换检验只花了7秒钟,我是笔记本i7-9700的cpu,睿频很低。
(6)将这么多结果中,显著的结果单独提取出来,形成data.table
mtpc.diffs.sig.dt <-
rbindlist(lapply(mtpc.diffs.list, function(x)
rbindlist(lapply(x, function(y)
y$DT[A.mtpc > A.crit, .SD[1], by=region]))))
DT就是data.table。
(7)
NBS network-based analysis
基于网络的NBS分析,也是一种常用的网络校正方法,DPABINet中有这个方法,matlab也有单独的包。这里也可以使用NBS方法。
在brain network中,以AAL模板举例,它有90个nodes,它里面的connections最大有4005个连接,所以某一个连接是否具有显著性,存在大量的假阳性。
由于边的数量这么多,如果选择FWE校正就会导致结果过小,丢失很多阳性结果。因此会倾向于使用FDR校正。而考虑到这些节点连接其实是有相互影响的,即不是独立分布的,因为FDR也不太合适。NBS就会成为一般通用选择。

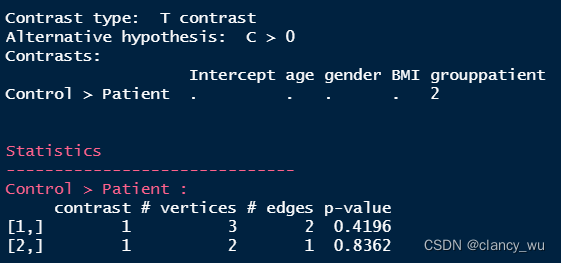

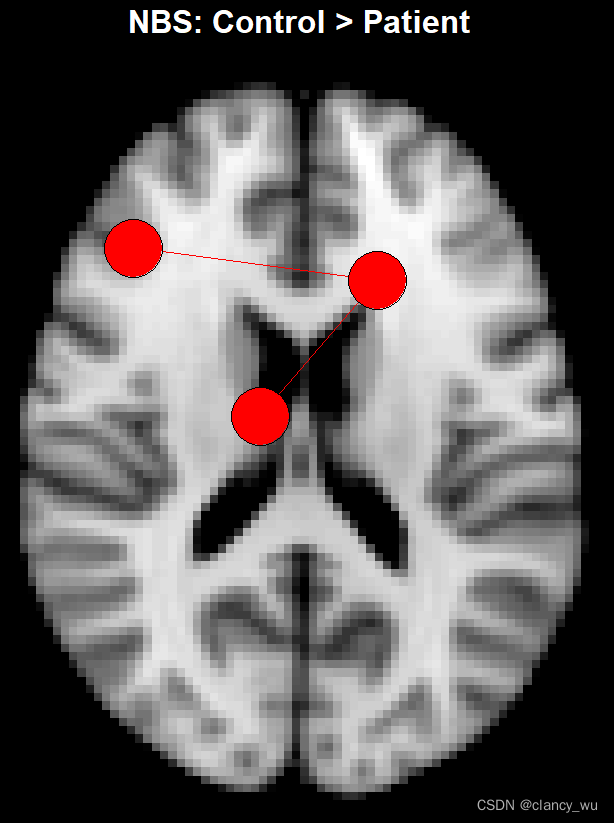
res.nbs的结果有2个,其实是permutation之后,排名靠前的cluster。然后我这个数据算的最大的cluster是3个点2个边,P是0.4,当然这个只是示例,如果P<0.05就是一篇文章了。
那么怎么查看属于哪个component呢,一种方法是查看它的p.mat,可以看到对应的P值。这里的plot其实就是叠加在模板上进行可视化了。
因为是示范,而且我的数据最小的P是0.4,所以我设置了alpha是0.5,这样就只有1个component显示,从上图可以看出,确实是3个点2个边。假如把alphas设置为0.9,按理说会有5个点和3条边,除非是有重叠的component,如下:
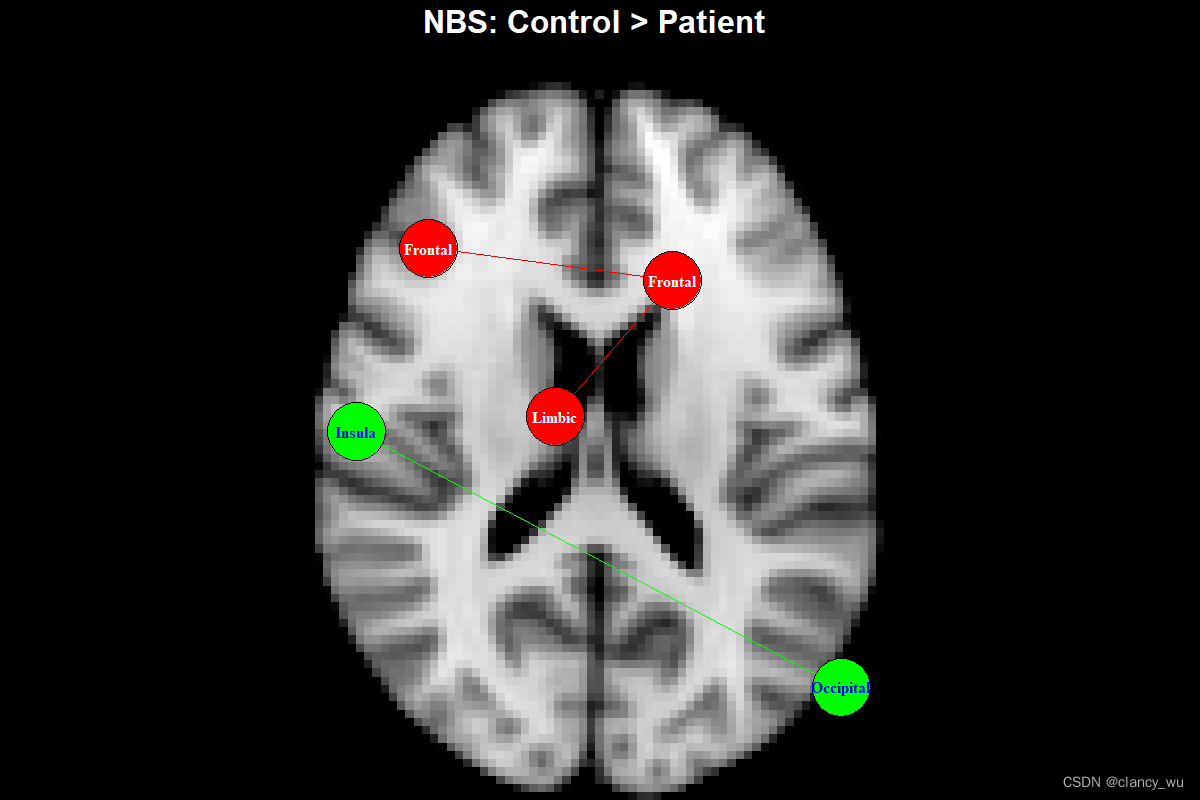
果然如我们所想。因为我用的是皮层模板,所以点都在皮层的沟回部分,然后因为这里的点的size太大,所以超出了图像边界,不过这个不重要。
作者将这个结果和matlab的NBS结果进行了对比,同样的数据,其T统计值都是一样的,而connect component有1个成分的差别,其他都一样,可能是由于NBS是按照T统计值排序component的,而这里是用P value去排序的,所以有了这么细微的差别,整体来说两者可以算是相同的。考虑到随机置换检验都会有种子点的差别和不同cpu的浮点计算差异,这种是可以接受的。
画图参数:
plot(x, plane = c("axial", "sagittal", "circular"),
hemi = c("both", "L", "R"), mni = TRUE, subgraph = NULL,
main = NULL, subtitle = "default", label = NULL, side = 1,
line = -2, adj = 0.025, cex = 2.5, col = "white", font = 2,
show.legend = FALSE, rescale = FALSE, asp = 0, ...)
Mediation analysis 中介分析
中介分析是用于检验自变量X对因变量Y(即X→Y)的影响是否由自变量通过影响中介变量M而对因变量Y(即X→M→Y)的产生影响。
感觉像是简单相关和偏相关的进一步分析。
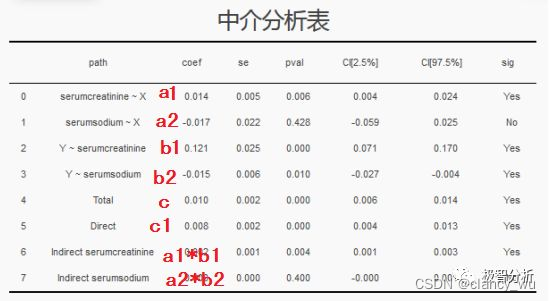
以知乎里极智分析作者写的文章为示例。假如要探究“年龄”(age,设为X)对“死亡”(DEATHEVENT,设为Y)的影响,看看年龄是否通过影响“血液中的血清肌酐水平”(serumcreatinine,设为A)、“血液中的血清钠水平”(serumsodium,设为B)而对死亡产生影响。建立中介分析模型,结果如下
- A与X有简单相关性(0),B与X没有简单相关性(1),Y与A有简单相关性(2),Y与B有简单相关性(3)
- 纳入A,B后,X与Y(total,4)有简单相关性;排除A,B后,X与Y(direct,5)有直接相关性(偏相关)
- 仅纳入A后,X与Y(6)有简单相关性;仅纳入B后,X与Y(7)有简单相关性
首先应用mediation analysis的前提就是X与Y有相关性。我们在临床中,会遇到一种情况,就是X与Y做cor.test,有结果,然后做pcor.test,就没有结果或者R变小了。这种情况就是因为confund factors中有某些indicators与Y直接相关,X其实是间接相关。遇到这种情况,我们就可以选定个别factors做中介分析。
其实中介分析很简单,就是不断的重复简单相关和偏相关来查看变量间关系。例如在上述例子中可以看出,A能影响XY,B只能影响Y,X与Y有简单相关和偏相关(说明X至少是影响Y的一部分原因或者全部原因)。
如果X与Y有简单相关,A与XY有简单相关,纳入A后X与Y偏相关不显著,那就说明A是完全中介作用,如果偏相关显著但是R值降低,就是部分中介作用。
因为要探究的是X与Y之间的具有中介效应的变量,而B与X不相关,所以直接排除B,而A的结果都符合预期,可以看出A是X与Y的中介变量,具有部分中介效应。
继续回到图论。
random graph, small world, rich-club
上述的图的研究方法,是从实际图出发来探究图的内在关系。有一些连接确定的图是可以这样的,例如社交网络。不过在核磁中,像functional network 和 structural covariance network,他们是通过相关性形成的图,这种会产生大量的虚假连接,导致它们的图表现出high level of clustering。那么为了解决这个问题,就要应用random graph方法,就是在相同degree distribution下随机生成N张图,random graph的特点是low level of clustering,因此可以缓解上述问题(当然只是缓解,无法做到完全克服)。
每生成1张random graph,就会有modularity, clustering coefficien, average path length, global efficiency, rich club coefficient这些指标,然后再把他们进行平均化比对。

random graph的过程还是比较漫长的,50个threshold的计算,花了近2小时(22:44 - 00:28, windows),如果在linux系统下进行超线程运行,应该会快蛮多。
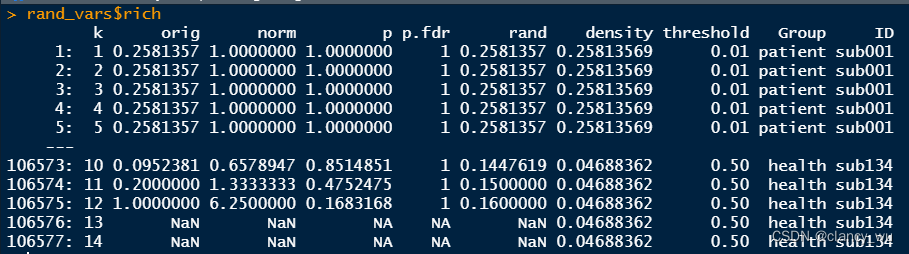
每个被试最高有1508个值,最低有1497个值,所以加起来产生了106577个值。这个跟每次计算出来的k值有关系。
small world就很符合预期,50个threshold X 71个被试 = 3550个值。
E.global等等指标也符合预期,50个threshold X 100 个随机图 X 71个被试。
上面介绍的是degree distribution 相同的random graph,其实还有更精确的一种随机方式,就是degree distribution 和 clustering相同的随机图,当然后者花费的时间会更长。(sim.rand.graph.clust & sim.rand.graph.par)
此外还有随机方差矩阵可以创建,random covariance metrics。
small-worldness
小世界属性,我们都知道,比较经典,即若网络具有高聚类性和短路径性,就说明它具有小世界属性(小小的世界,四通八达,地球村)。研究小世界属性的经典方法就是生成一系列等效网络来研究(equivalent network),就是我们上述所说的步骤。
One popular method for studying small-world networks is to use an equivalent network model to generate other similar instances of the class of systems under study.
σ
=
C
/
C
r
a
n
d
L
/
L
r
a
n
d
\sigma = \frac{C / C_{rand}}{L / L_{rand}}
σ=L/LrandC/Crand
然后Telesford认为
σ
\sigma
σ将真实网络聚类与随机网络聚类的对比会产生异常值,使得部分不具备小世界属性的网络表现出了小世界属性。如下:


因此他提出了
ω
\omega
ω,
ω
\omega
ω的范围在【-1,1】之间,而当其处于【-0.5,0.5】时,就认为其具有小世界属性,当值位于两端时,表示为其是pure lattice 或者 random network。
boostrapping and permutation test
boostrapping test 和 permutation test 是两种检测两组图有无差异的方法,即可以在node-level或者是edge-level去比较两组有无差异。
















 2万+
2万+











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











