







Yuansong Zeng, Zhuoyi Wei, Fengqi Zhong, Zixiang Pan, Yutong Lu, Yuedong Yang, A parameter-free deep embedded clustering method for single-cell RNA-seq data, Briefings in Bioinformatics, Volume 23, Issue 5, September 2022, bbac172, parameter-free deep embedded clustering method for single-cell RNA-seq data | Briefings in Bioinformatics | Oxford Academic
代码:
摘要
单细胞核糖核酸(RNA)测序数据中的聚类分析被广泛用于发现细胞的异质性和状态。尽管已经开发了许多用于单细胞RNA测序分析的聚类方法,但大多数这些方法都需要提供聚类的数量。然而,事先知道细胞类型的确切数量并不容易,而且经验决定也不总是可靠的。本文开发了ADClust,一种用于单细胞RNA测序数据的自动深度嵌入聚类方法,可以在不需要预先定义聚类数量的情况下准确地对细胞进行聚类。具体而言,ADClust首先通过预训练的自编码器获得低维表示,并使用这些表示将细胞聚类成初始微聚类。然后,通过统计测试比较这些聚类,将相似的微聚类合并成更大的聚类。根据聚类结果,更新细胞表示,使每个细胞被拉向其分配聚类和相似聚类的中心,同时将细胞分开以保持聚类之间的距离。这是通过共同优化精心设计的聚类和自编码器损失函数来实现的。此合并过程持续进行直至收敛。ADClust在12个真实的单细胞RNA测序数据集上进行了测试,并显示在聚类性能和确定聚类数量的准确性方面优于现有方法。更重要的是,我们的模型为大型数据集提供了高速和可伸缩性。
ADClust方法
ADClust模型由两个模块组成:自编码器和聚类优化模块。自编码器旨在学习细胞的深度嵌入表示,而聚类优化模块则使用学习到的嵌入表示对细胞进行聚类。
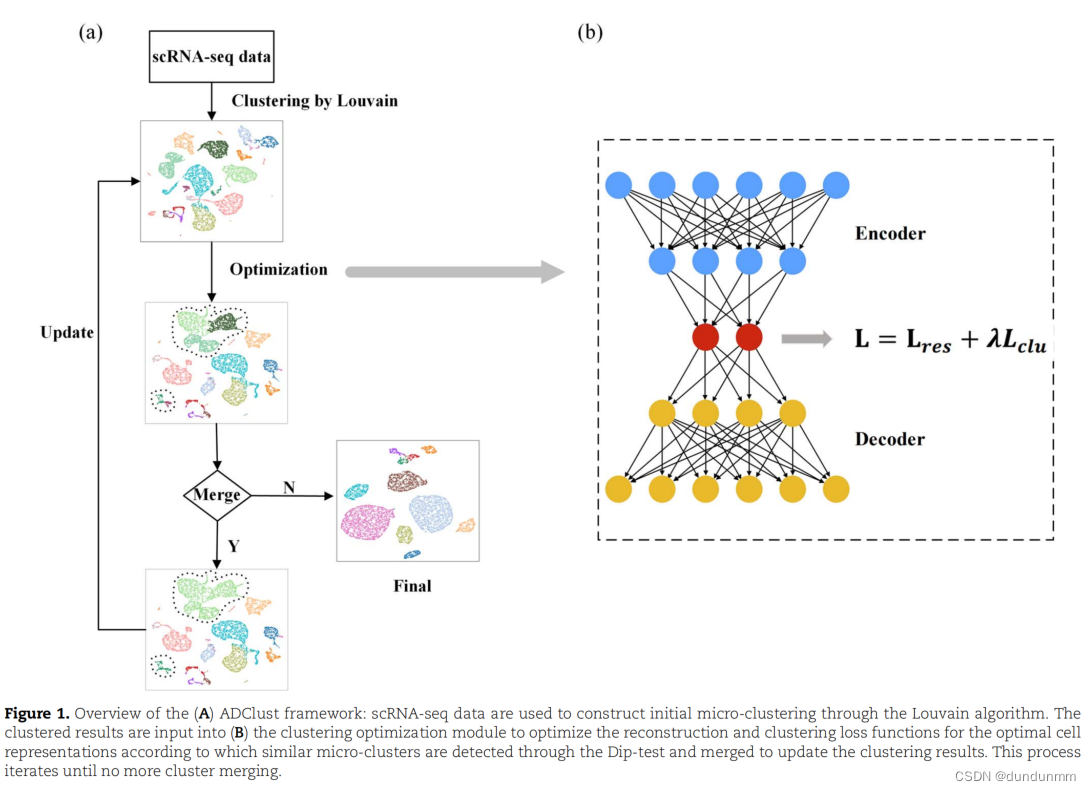
自编码器模块
用于将输入的单细胞RNA测序基因表达数据 X 嵌入到低维空间中。自编码器是一种无监督神经网络,由编码器和解码器模块组成。编码器试图将输入数据嵌入到潜在空间中,而解码器试图将嵌入的数据重构回其原始空间。因此,通过最小化重构损失 Lres,自编码器可以有效地学习有用的低维潜在空间,计算公式如下:

其中,dec() 和 enc() 分别表示编码器和解码器函数。enc(x) 是对个体细胞基因表达 x 的学习嵌入表示。
聚类优化模块
基于学习到的嵌入表示,通过Louvain算法[36]将细胞聚类成丰富的初始微聚类。然后,通过Dip检验比较微聚类之间的差异,并通过精心设计的聚类损失函数合并相似的微聚类。
Dip检验和Dip得分
Dip检验是一种测量多模态性的统计检验,最早由J. A. Hartigan和P. M. Hartigan于20世纪80年代开发[29]。该检验检测两个聚类的样本(细胞)集,并输出一个Dip得分 ∈[0,1] 作为样本集单峰性的概率。Dip得分较大的两个微聚类表示它们之间的结构相似性较高,在本文的方法中将被合并。
聚类损失
通过聚类损失Lclu,将相似的微聚类在自编码器的嵌入空间中拉近,该损失最初在[37]中使用:
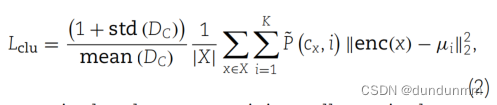
其中,cx 是包含细胞 x 的聚类,μ 是K个聚类的中心,![]() 反映了通过Dip检验估计的聚类中心cx之间的归一化Dip得分。‘mean’ 和 ‘std’ 是聚类对之间距离DC集的均值和标准差。
反映了通过Dip检验估计的聚类中心cx之间的归一化Dip得分。‘mean’ 和 ‘std’ 是聚类对之间距离DC集的均值和标准差。
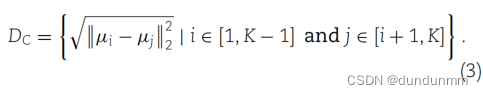
直观来说,模型通过减小细胞与其分配中心之间的距离来最小化损失。同时,细胞也会根据与聚类i的相似性而被拉向其相似的聚类,拉力的强度取决于与聚类i的相似性。因此,如果具有较大Dip得分的聚类是相似的,模型将减小聚类之间的距离。这个过程将把相似的微聚类拉在一起。通过除以均值,可以防止自编码器仅减小嵌入尺度以最小化损失Lclu。还应用std来阻止模型减小尺度并同时将单个聚类推远。最后,以端到端的方式优化ADClust,使用以下损失函数:

其中λ是平衡聚类损失贡献的超参数。在本研究中,为所有数据集设置λ = 1。
合并过程
如果两个聚类的相应Dip得分大于Dip得分阈值,则将合并它们。这两个合并的聚类中的细胞将被分配相同的细胞标签,并且这些细胞的新中心将计算如下:
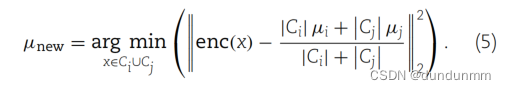
根据新的中心μnew,需要更新Dip得分矩阵P和 ˜P。如果P中仍有大于阈值的Dip得分,则重复合并过程。合并过程完成后,继续优化模型并合并聚类。这个过程会一直持续,直到没有Dip得分大于阈值的情况。
实验
实验数据
实验数据分为了两部分:模拟数据和真实数据
虚拟数据
使用Splatter软件包[31]生成了模拟单细胞RNA测序数据集。为了模拟具有不同聚类信号强度的单细胞RNA测序数据集,生成了不同de.facScale值的数据集,其中de.facScale的取值范围为{0.2, 0.25, 0.3, 0.35, 0.4}。较高的de.facScale值代表着更强的聚类信号,对应于更容易的数据集。每个数据集包含2000个细胞和2000个基因。参数dropout.mid设置为2(固定的失活率约为45%)。按照研究[32]的方法设置参数group.prob为{0.1, 0.15, 0.25, 0.5},以便每个模拟数据集包含四组不同比例的数据。对于其他参数,使用默认值。使用不同的随机种子重复生成数据集五次,并展示平均结果。
真实数据
这些数据集涉及不同的生物过程和各种组织,包含从几千到几万个不同规模的细胞,这些细胞来自各种单细胞RNA测序技术。每个数据集都使用Seurat提出的标准流程进行预处理。具体而言,通过“NormalizeData”函数对基因表达进行了归一化处理,使用默认参数“LogNormalize”(对于模拟数据为“RC”),缩放因子为10,000。然后,基于归一化矩阵,通过“FindVariableFeatures”函数选择了前2000个高度变异的基因。
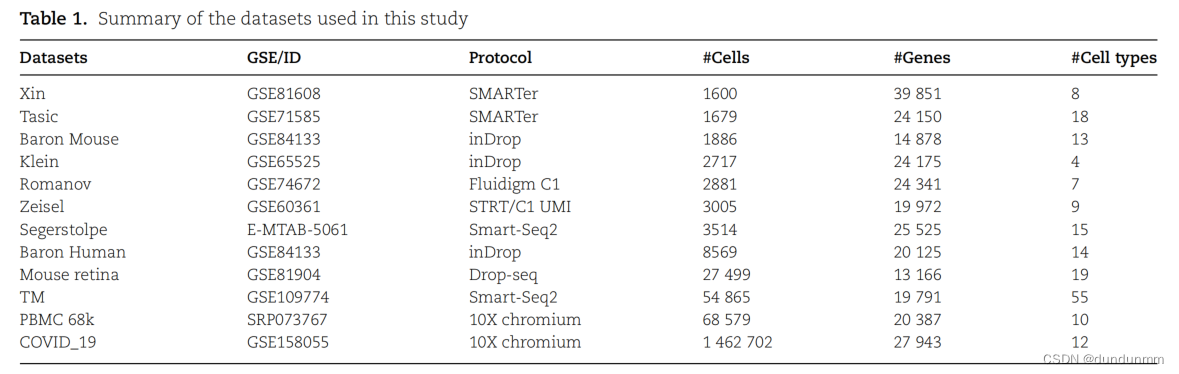
实验结果
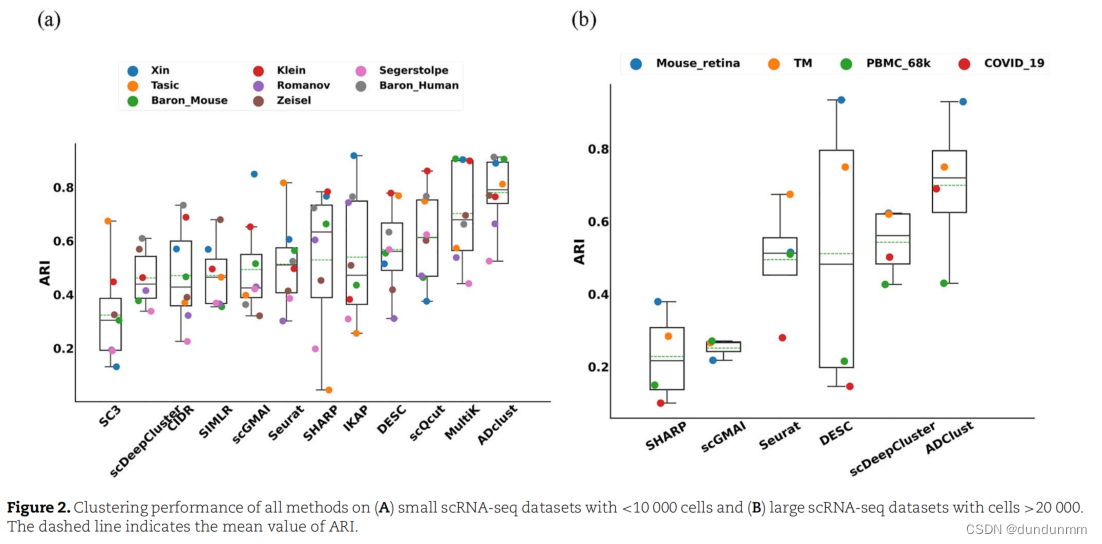
这篇文章与之前几篇分享的单细胞RNA测序数据聚类不同的点在于,其将注意力放于细胞类簇个数的自动学习上。
无参聚类一直是聚类领域的研究重点和难点,目前深度的方法很少,是一个值得考虑研究的方向。















 649
649

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









