







1.amber输出文件:(格式不唯一)
拓扑文件parm: compw.prmtop
轨迹文件:prod.mdcrd
2.打开vmd输出最后一帧,保存为PDB格式
vmd
2.1加载拓扑文件
**
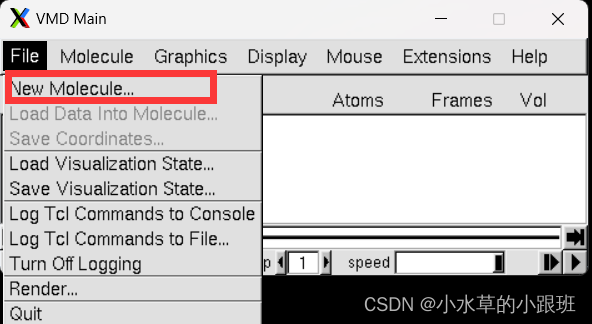
点击file-New Molecule
**
‘’
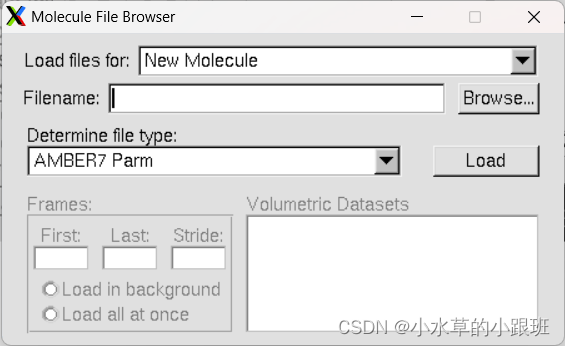
将Determine file type 改为正确类型
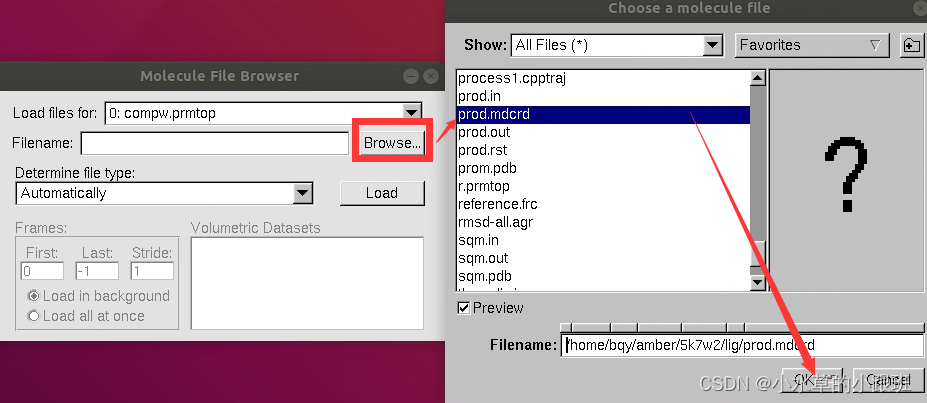
首先加载拓扑文件,点击Browse
‘’
‘’
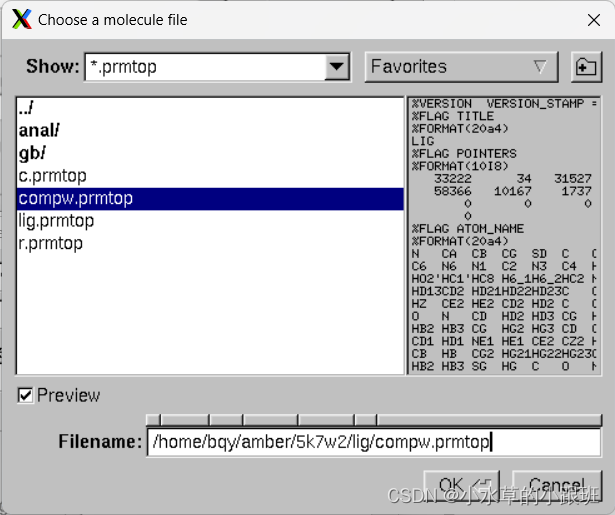
选择输出的拓扑文件
‘’
‘’
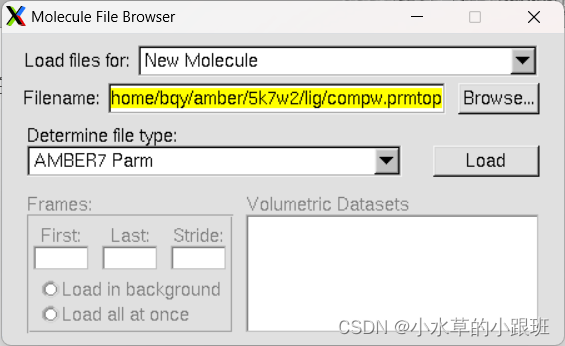
点击Load
‘’
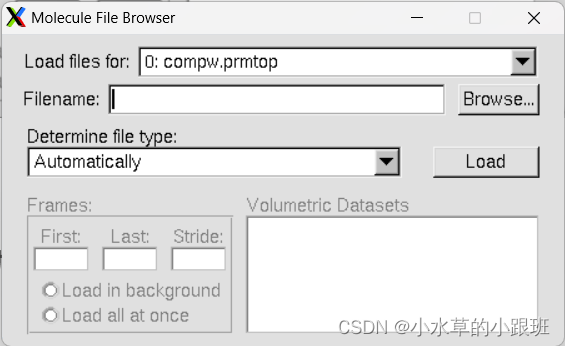
2.2 加载轨迹文件
‘’
上一步点击Load后界面如图,不要退出
选择正确的Determine file type:NetCDF(AMBER,MMTK)
‘’
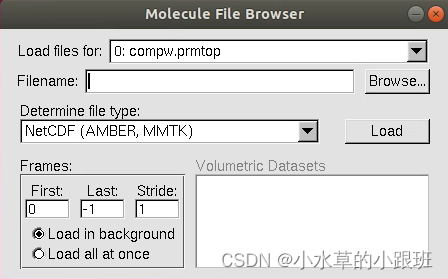
‘’
再点击Load即可
‘’
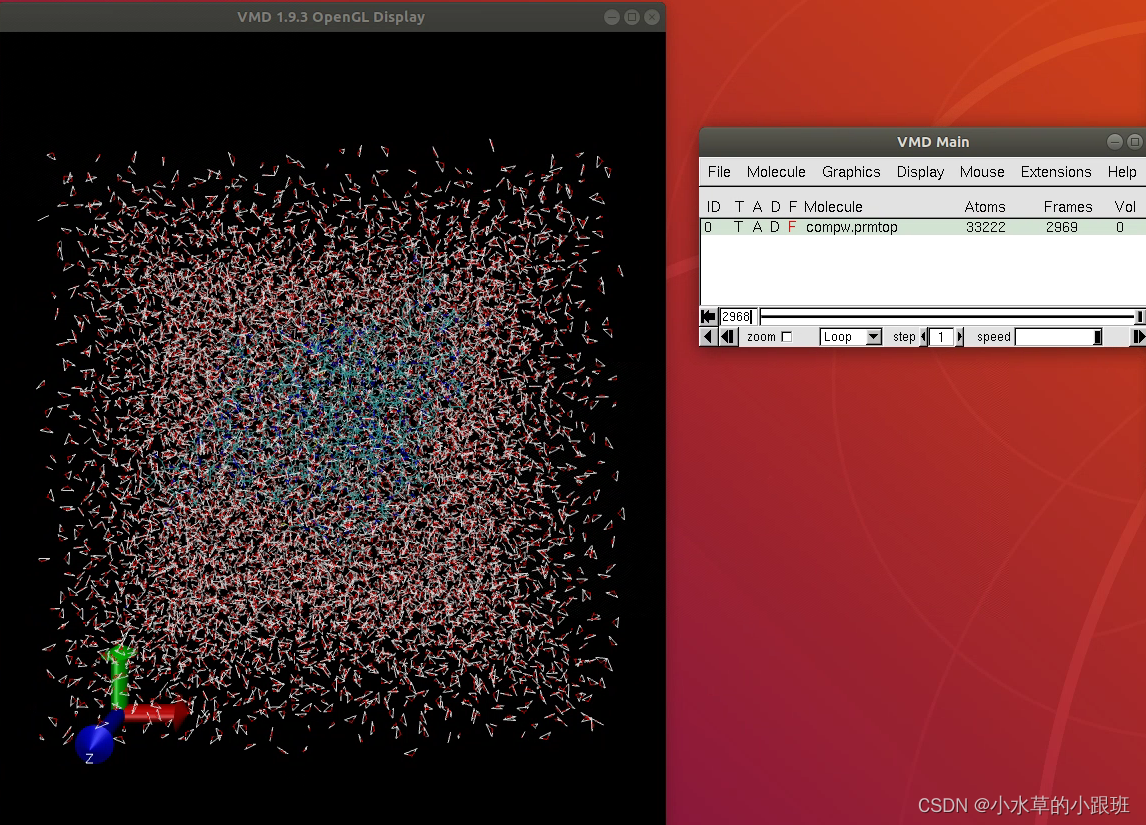
2.3输出最后一帧并保存为pdb格式
‘’
完成上两步后
‘’
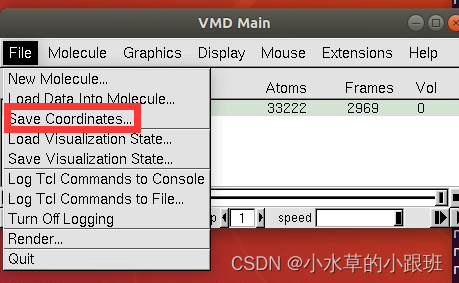
‘’
File-Save Coordinates
‘’
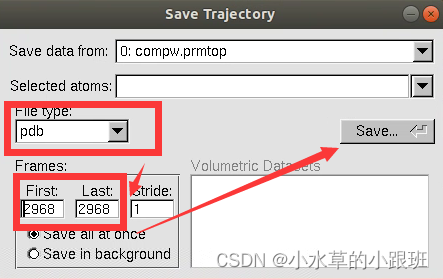
‘’
最后再按照提示选择保存的路径即可
‘’
3.MD轨迹分析
3.1新建脚本文件,文件名.后缀:mdana.cpptraj
3.2写入脚本文件
文件内容:
parm compw.prmtop
trajin prod.mdcrd
rmsd all first :1-24&!@H= out rmsd-all.agr mass
解释:第一行为加载拓扑文件, parm不可修改,要修改后面的文件名
第二行加载轨迹文件 ,trajin不可修改,需要修改后面的文件名
第三行冒号前:使用第一帧为基准
第三行冒号后:计算1-24号残基,并且不计算H的部分,输出文件为rmsd-all.agr
3.3 执行脚本
记得要安装amber,module load amber
cpptraj -i process1.cpptraj
3.4打开输出文件
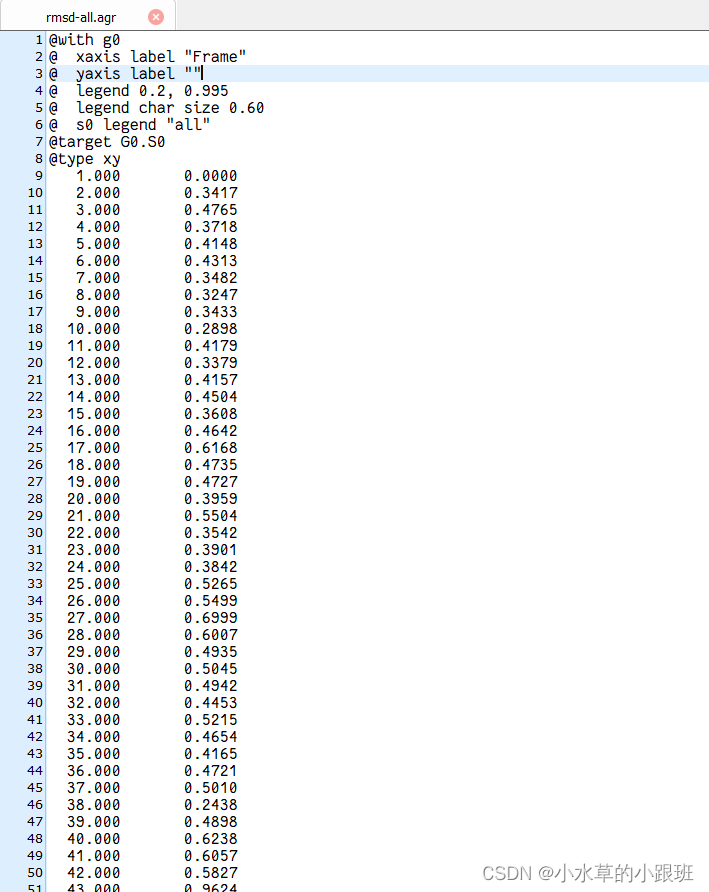
下图为输出的数据,可根据输出数据用自己的工具绘制轨迹图
例如python绘图
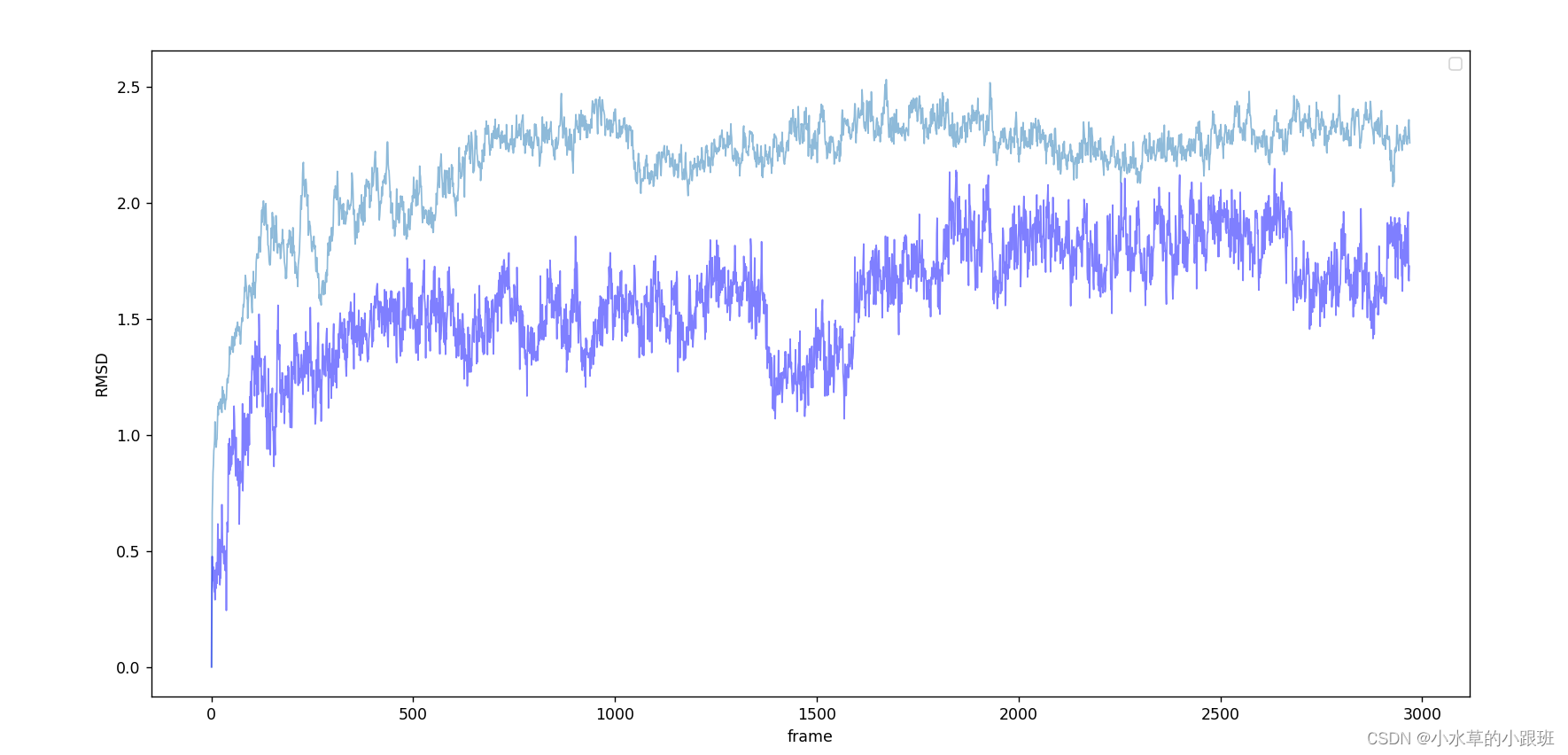
坐标自行修改即可















 3855
3855











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









