






分歧时间是当前进化分析的一个热点,以某几个特定类群的化石时间作为校正,然后通过基因序列间的分歧程度以及分子钟来估算物种间的分歧时间,同时估算系统发育树上其它节点的发生时间,从而推断相关类群的起源和不同类群的分歧时间。目前,采用依据BI树和ML树估计物种分歧时间的程序很多,例如R8S、MCMCTREE、MULTIDIVTIME、BEAST、MEGA等,通过不同的策略将化石时间信息整合到一个系统发育树中,从而计算得到Divergence time Tree。
进化速率(Evolutionary rate)指分子的进化速率(蛋白质或核酸等大分子中的氨基酸或核苷酸在一定时间内的替换率),也可指某一类群的物种分化速率,甚至可指特定性状的进化速率。因此,笼统地说,进化速率指在进化过程中,单位时间内发生的改变。分子钟的研究与分子进化速率相关。
分子进化速率有针对病毒进化速率研究或非编码序列的nucleotide substitution速率,也有针对编码序列的密码子同义替换速率与非同义替换速率。
一、基本概念
1.分子钟概念
分子钟的概念源于对蛋白质的研究。Zuckerkandl and Pauling (1962)比较几种动物的血红蛋白、细胞色素C的序列后注意到:这些蛋白质的氨基酸取代速率在不同的种系间大致相同,即分子水平的进化存在恒速现象。Zuckerkandl and Pauling (1965)提出将1962年提出的概念命名为分子钟,分子水平进化存在一个“时钟“, 也即进化速率是近似恒定的。因此,分子钟假设的成立条件是对于给定的任意大分子(DNA序列或者蛋白质序列)在所有进化谱系中有近似恒定的进化速率,如在一个进化分支上所聚集的突变数与该分支的独立进化时间长度成正比,其替代速率在进化过程中近似保持一个恒定的数值。
2.分子钟应用面临的问题
分子钟假说成立的条件DNA或者蛋白质序列的替代速率是恒定的。20世纪80年代以来,随着DNA序列数据快速积累,大量的证据表明:在长期进化过程中,很多类群的绝大多数基因或蛋白质的序列替换速率根本不符合分子钟假说。对于蛋白质序列,在物种适应辐射过程中,其进化速度可能会大大加快。因此,以蛋白质为基础的恒定进化速率并非理想的分子钟;对于核酸分子,不同基因的分子钟速率不同;并且同一基因在不同的生物类群间可能有显著差异,因此同一基因的分子异速进化现象是显而易见的。目前分子钟面临的一些挑战也主要与分子异速进化相关,由于分子异速进化的存在,特别是同一级基因在不同生物类群的进化速率可能有显著的差异,这给应用同一分子钟来重建物种系统发育关系及估算物种分歧年代带来了困难,这是分子钟在应用上面临的有一个挑战。
3.分子钟的类型
在BEAST2中,分子钟模型的模型可选项有四个包括:-Strict Clock(严格分子钟); -Relaxed Clock Exponential(松散指数分子钟); -Relaxed Clock Log Normal(松散对数分子钟); -Random Local Clock(随机本地分子钟)。
4.分子钟校正
分子进化是生物分子层次上的进化,分子系统学是从生物大分子(蛋白质、核算)的信息推断生物进化历史,或者说重建系统发育关系,并以系统树的形式表示出来。系统发生树的枝长仅代表每位点的碱基替代数或者遗传距离。基于生物遗传距离所建立的分子进化速率,使得分子钟方法在分子系统学中的应用使得利用片段估算物种的起源和分化时间成为可能,并可用于进一步推测不同生物类群在进化历史上的分歧时间。在速率恒定的假设下,遗传距离是时间的线性函数,为了将遗传距离转化为分歧时间,至少需要一个能够提供时间信息的标定点(calibration point)。常用的校准信息可以分为:(1)已知的碱基替代速率;(2)化石校准点;(3)生物地理事件校准点;(4)二次校准点
5.中性理论
受达尔文自然选择学说影响,当分子时钟刚提出的时候,人们认为决定氨基酸替换或者碱基替换的关键性因素是自然选择。如果分子时钟成立,那么也就意味着适应性替换在物种的发生是恒定的。但是适应性变异的发生和固定受到很多因素的影响,比如选择压力大小、群体效应大小、突变率等等,这些因素在各个物种中差异很大。但是这些因素作用下的适应性变异为什么会是很定的呢?为此,Kimura在1968年提出了另一个理论,认为如果一个突变没有带来表型差异,那么它在该群体中的命运则完全受各种随机因素的影响。也就是”中性理论“。
二、mcmctree估算物种分歧时间
1.第一步构建物种树
使用单拷贝直系同源基因四倍简并位点或者线粒体基因,通过IQTREE或者Mrbayes等软件构建物种的系统发育树。这里就不详细讲建树流程了。
2. 获取化石分歧时间
这一步还是很关键的,只有根据标定的化石点才可以估算出树中每个物种的分化时间。可以根据已发表的文章获得化石点,可以标定2-3个化石点,也可以5-10个化石点,根据物种树的规模来看。
假如在已发表的文章中没有找到可用的化石点,那么这里有一个很好用的物种分化时间查询工具 - TimeTree
TimeTree通过汇总大量的原始文献,包括大量的人工核实,估计出不同物种的最近共同祖先,并构建出整个所有可及物种的进化树。目前TimeTree来到了第五个版本,包含了4000多个相关的研究,以及近14万个物种。通过网页版或者手机APP版可以方便查询任意两个物种的分化时间,或者多个物种的进化树。我们以黑腹果蝇Drosophila melanogaster, 冈比亚按蚊Anopheles gambiae为例。
网站链接:http://www.timetree.org/
(1)单个物种的进化过程
我们在网页中输入黑腹果蝇:
然后我们可以看到整个黑腹果蝇的进化历程,包括细胞的形成,细胞核的形成……双翅目的形成……果蝇亚组的形成,从数十亿年前到今天的整个过程。
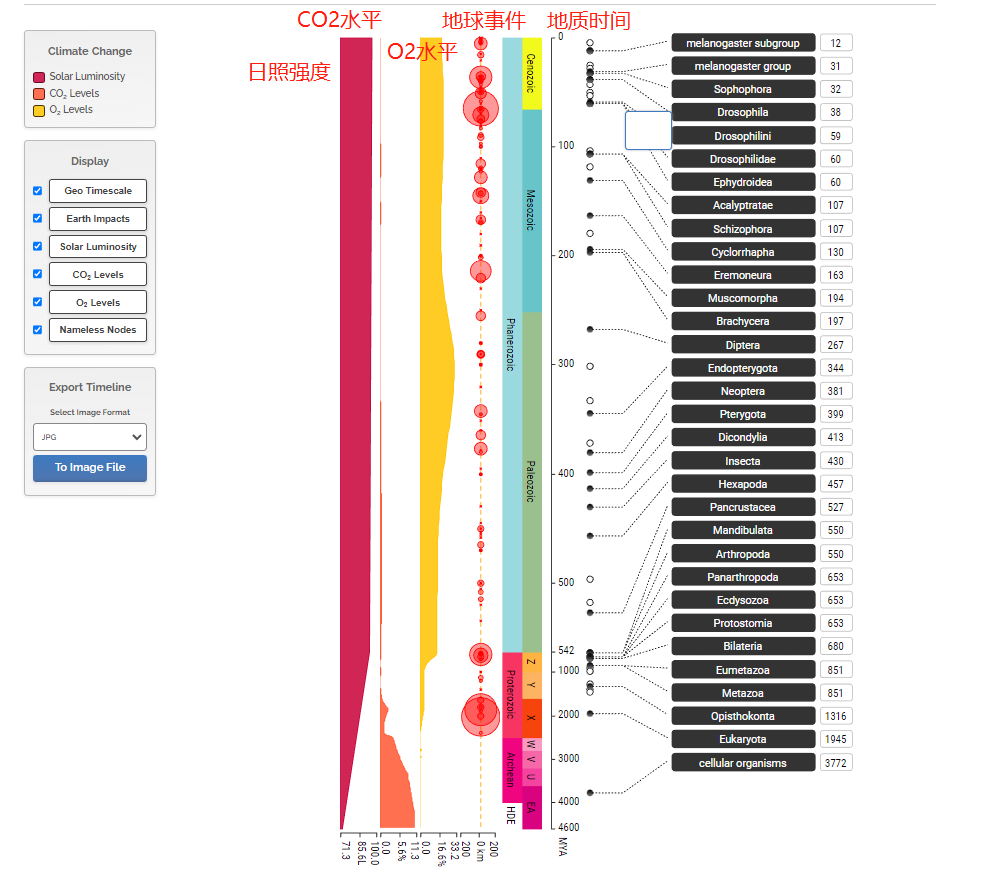
有意思的是,整个时间线不仅包括了物种的进化和形成,还包括了非生物因素的变化,比如日照强度,二氧化碳水平,氧气水平。同时,还有一些地球(宇宙)事件的影响,比如彗星的撞击等。比如点击65mya左右的大红圈,会显示墨西哥的彗星撞击。
(2)两个物种的分化时间
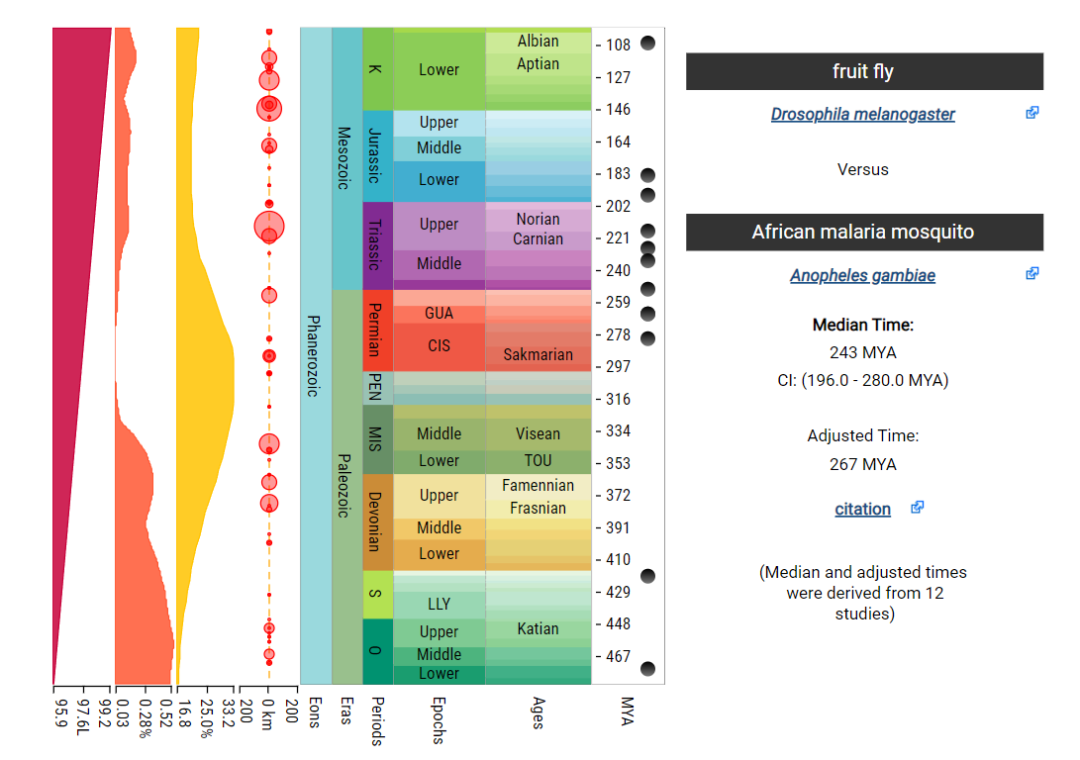
在如下网页中输入黑腹果蝇和冈比亚按蚊。
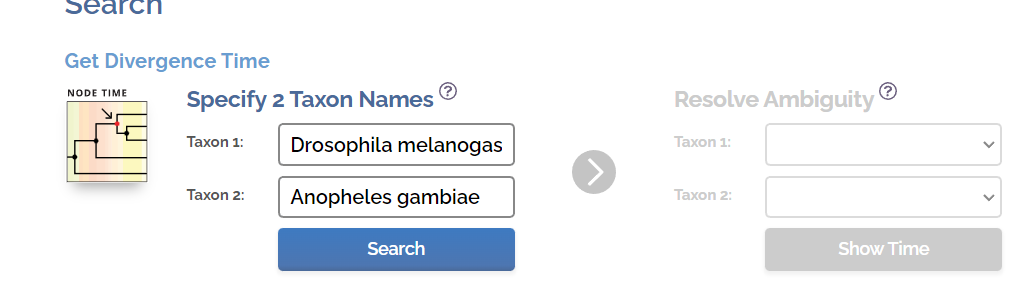
根据12个不同的研究,估计出黑腹果蝇和冈比亚按蚊的中位分化时间大约在2.43亿年前, 95%CI(1.96, 2.80)。
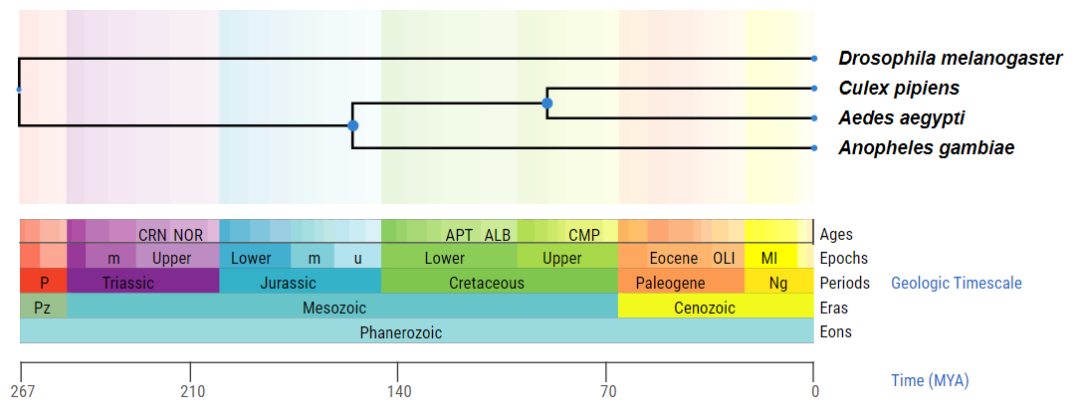
(3)多个物种的进化树
可以输入某个物种群组,把该群组中的所有物种的关系都显示出来。或者上传一个自定义的物种列表。比如我们此处随意输入4个物种:Drosophila melanogaster (黑腹果蝇) Anopheles gambiae (冈比亚按蚊)Aedes aegypti (埃及伊蚊) ulex pipiens (淡色库蚊)
3.估算物种分歧时间
step 1: 估计替换速率
输入文件:newick格式的系统发育树(不带分支长度)、多序列比对文件、baseml控制文件
13 1(((((((Ba,((Hm,(Mc,Kin)),Dp)),(Cc,Cn)),Pr),La),Pp),(Bm,Ha)),Dm)'@2.72';
seqfile = allSingleCopyOrthologsAlign.4Dsite.fastreefile = species.tree0outfile = mlb * main result filenoisy = 3 * 0,1,2,3: how much rubbish on the screenverbose = 1 * 1: detailed output, 0: concise outputrunmode = 0 * 0:user tree; 1:semi-automatic; 2:automatic* 3:StepwiseAddition; (4,5):PerturbationNNImodel = 7 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85, 5:T92, 6:TN93, 7:REV* 8:UNREST, 9:REVu; 10:UNRESTuMgene = 0 * 0:rates, 1:separate; 2:diff pi, 3:diff kappa, 4:all diffclock = 1 * 0:no clock, 1:clock; 2:local clock; 3:CombinedAnalysisfix_kappa = 0 * 0: estimate kappa; 1: fix kappa at value belowkappa = 2 * initial or fixed kappasfix_alpha = 0 * 0: estimate alpha; 1: fix alpha at value belowalpha = 0.5 * initial or fixed alpha, 0:infinity (constant rate)ncatG = 5 * # of categories in the dG, AdG, or nparK models of ratesfix_rho = 1 * 0: estimate rho; 1: fix rho at value belowrho = 0 * initial or fixed rho, 0:no correlationMalpha = 0 * 1: different alpha's for genes, 0: one alphanparK = 0 * rate-class models. 1:rK, 2:rK&fK, 3:rK&MK(1/K), 4:rK&MKgetSE = 1 * 0: don't want SEs of estimates, 1: want SEsRateAncestor = 0 * (0,1,2): rates (alpha>0) or ancestral statesmethod = 0 * Optimization method 0: simultaneous; 1: one branch a timeSmall_Diff = 0.5e-6cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?fix_blength = 0 * 0: ignore, -1: random, 1: initial, 2: fixed
运行
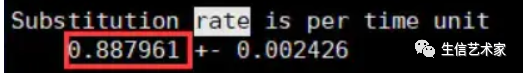
baseml baseml.ctl输出文件中,会得到替换速率,用于mcmctree运算。
step 2: 估计Gradient and Hessian of Branch Lengths
输入文件:newick格式的系统发育树(不带分支长度,带有多个化石校正时间)、多序列比对文件、mcmctree控制文件
13 1(((((((Ba,((Hm,(Mc,Kin))'B(58,84)',Dp)),(Cc,Cn)),Pr)'B(79,118)',La),Pp)'B(76,146)',(Bm,Ha)'B(100,122)'),Dm)'B(217,314)';
seed = -1seqfile = allSingleCopyOrthologsAlign.4Dsite.fastreefile = species.tree1outfile = out_usedata3ndata = 1usedata = 3 * 0: no data; 1:seq like; 2:normal approximationclock = 3 * 1: global clock; 2: independent rates; 3: correlated rates* RootAge = '<10' * constraint on root age, used if no fossil for root.model = 7 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85alpha = 0.5 * alpha for gamma rates at sitesncatG = 5 * No. categories in discrete gammacleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?BDparas = 1 1 0 * birth, death, samplingkappa_gamma = 6 2 * gamma prior for kappaalpha_gamma = 1 1 * gamma prior for alphargene_gamma = 1 2.288 * gamma prior for rate for genes ### 1/替换率sigma2_gamma = 1 4.5 * gamma prior for sigma^2 (for clock=2 or 3)finetune = 1: 0.06 0.5 0.006 0.12 0.4 * times, rates, mixing, paras, RateParasprint = 1burnin = 500000sampfreq = 5000nsample = 20000*** Note: Make your window wider (100 columns) when running this program.
运行
mcmctree mcmctree3.ctlstep 3: 估算物种分歧时间
输入文件:newick格式的系统发育树(不带分支长度,带有多个化石校正时间)、多序列比对文件、mcmctree控制文件、in.BV(由上一步分析获得)
mv out.BV in.BVseed = -1seqfile = allSingleCopyOrthologsAlign.4Dsite.fastreefile = species.tree1outfile = out_usedata2ndata = 1usedata = 2 * 0: no data; 1:seq like; 2:normal approximationclock = 3 * 1: global clock; 2: independent rates; 3: correlated rates* RootAge = '<10' * constraint on root age, used if no fossil for root.model = 7 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85alpha = 0.5 * alpha for gamma rates at sitesncatG = 5 * No. categories in discrete gammacleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?BDparas = 1 1 0 * birth, death, samplingkappa_gamma = 6 2 * gamma prior for kappaalpha_gamma = 1 1 * gamma prior for alphargene_gamma = 1 2.288 * gamma prior for rate for genes ### 1/替换率sigma2_gamma = 1 4.5 * gamma prior for sigma^2 (for clock=2 or 3)finetune = 1: 0.06 0.5 0.006 0.12 0.4 * times, rates, mixing, paras, RateParasprint = 1burnin = 500000sampfreq = 5000nsample = 20000*** Note: Make your window wider (100 columns) when running this program.
运行
nohup mcmctree mcmctree2.ctl >mcmctree.ctl.log 2>&1 &;检测运行结果
最直接的检测方法是:分别使用不同的Seed值进行mcmctree或infinitesites进行两次或多次分析,然后比较两个结果树是否一致,实际就是比较树文件中各内部节点的Height值(分歧时间 / Posterior time)。计算各枝长总的偏差百分比,当偏差百分比低于0.1%,则认为两次结果非常吻合,差异低于0.1%,认为达到收敛。此外,还可以使用Tracer分析mcmc.txt文件,检测其ESS值,一般认为该值高于200,则可能达到收敛。该方法可用于辅助检测。最后,若不收敛,则需要提高burnin、nsample值,重新运行程序。
三、MEGA-X估算物种分歧时间
MEGA-X中我们可以通过「分子钟」来追溯生物进化的历程,接下来我们来试一下用MEGA-X的RelTime方法来推断一下进化时间,首先在推断前需要准备的文件有以下几个:可用于推断分化时间的系统发育树(注意这里的发育树上需要有外群以及校准点)的序列文件以及Newick格式的树文件。
第一步 :打开MEGA-X ------ CLOCKS ------ Compute Timetree ------ RelTime-ML
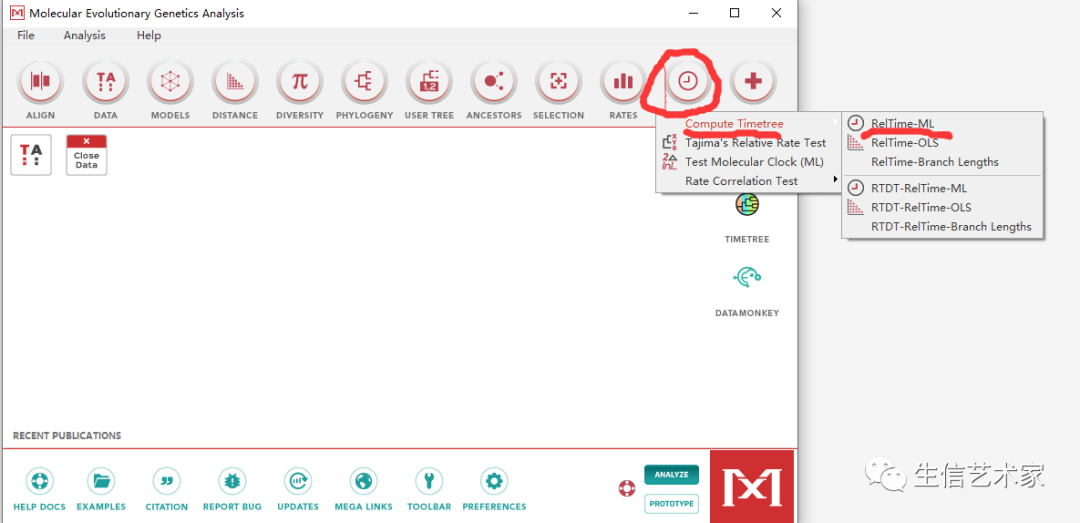
第二步 :先设置前三个输入(1)序列文件fasta格式 (2) 物种树文件 .nwk或者.tre格式 (3)设置外群
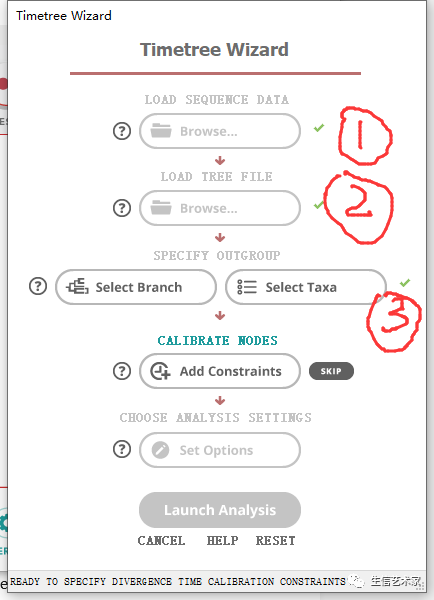
第三步 :标定化石点
在CALIBRATE NODES这,点Add Constraints就会进入下面的界面
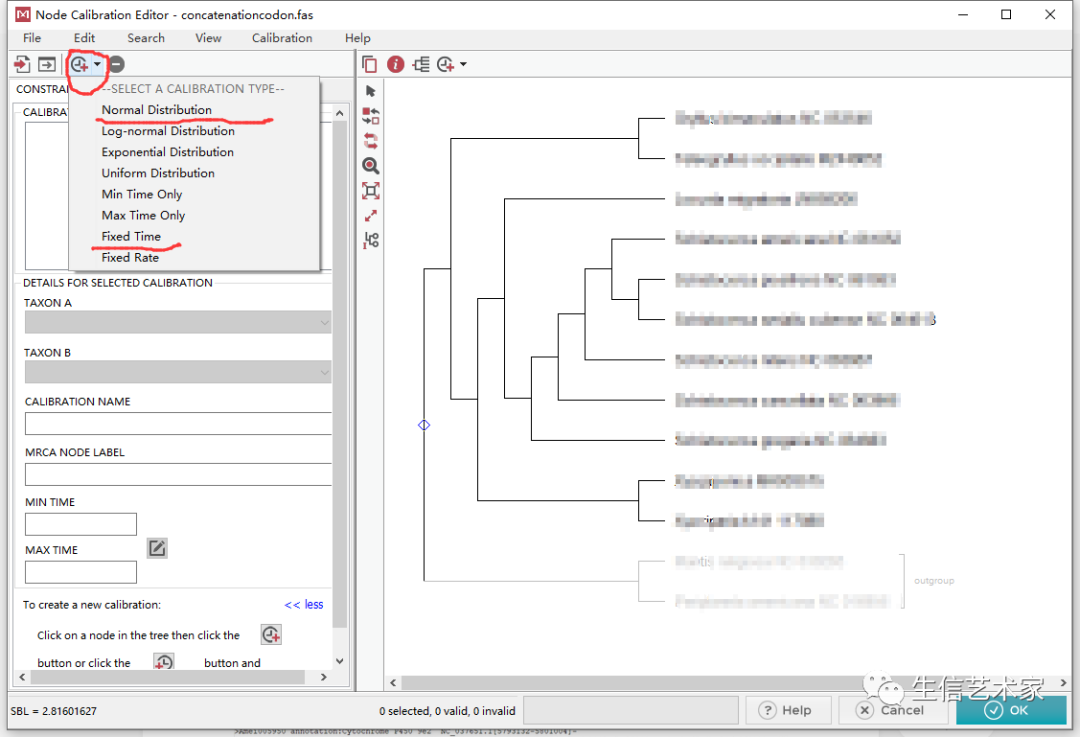
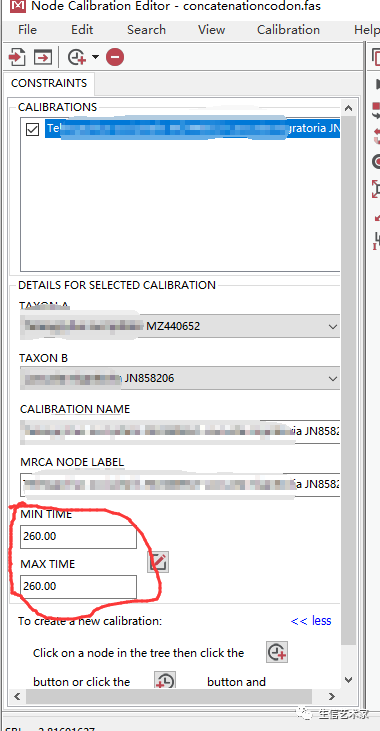
这里可以添加多个化石点,根据Timetree查询到的化石点就可以用,添加好了后点ok就行。
第四步 :设置可选参数
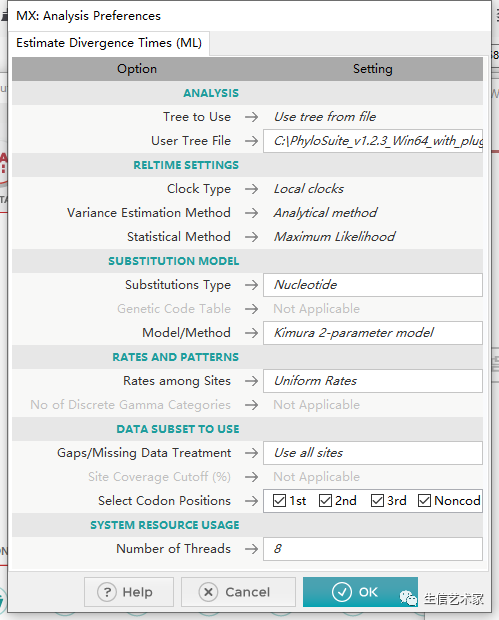
根据ML树计算分歧时间,一般默认参数就行,直接点击OK
第五步 :点击分析
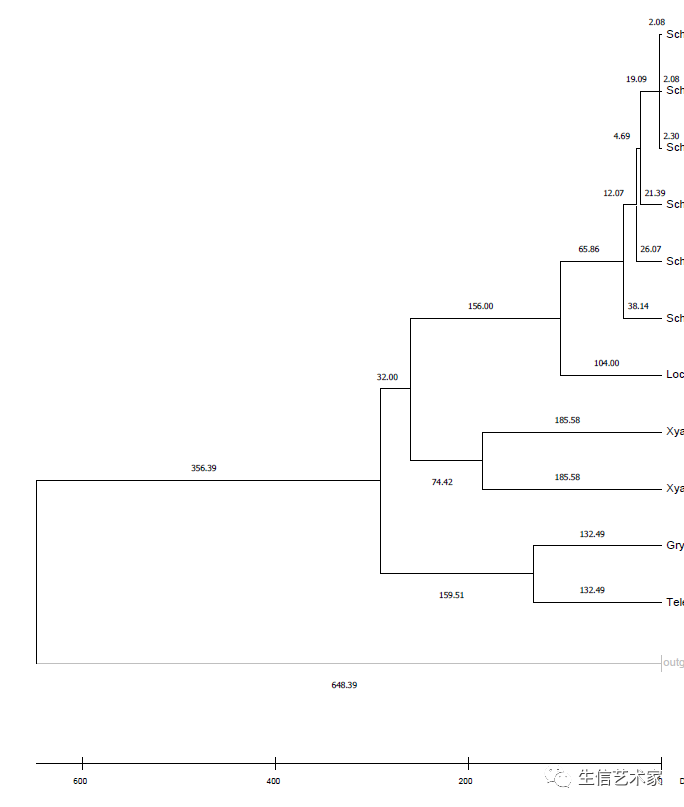
这里就会得到一个带有分歧时间的树,也可以显示每个节点的时间。
四、分子进化速率计算
进化速率,实际存在两种不同的东西
第一种就是对非编码区域来说,核酸的替代的速率。当然编码区域也可以用核酸替代速率来表示进化速率。这一种算法相对简单,单位就是nucleotide substitution per Myr。在病毒进化速率计算,非编码序列的进化速率计算时通常使用这种方法。
第二种就是对蛋白质编码基因来说,一般是密码子的同义替换率与非同义替换率来表示进化速率,以及通过dN/dS来表示选择压力的大小。可以通过PAML 中的codeml来计算。
1.核酸的替代的速率
这个计算起来很简单,根据这篇文章中的方法来计算
By dividing the pairwise nucleotide distances between Prolaupala and Laupala elements within each cluster by 10 million years (Myr) (twice the divergence time between Laupala from Prolaupala estimated from biogeography) (18), we obtain an estimate of the absolute rate of point substitution. The average over all clusters is 3.8 * 10-3 nucleotide substitution per Myr, which is almost four times lower than the 15 * 10-3 nucleotide substitution per Myr estimated in Drosophila (19).
公式就是 进化速率 =(P-distance) / (twice the divergence time)
那么具体怎么计算呢,不知道有没有专门的软件,这个可以自己写个代码算。
你先要获得一个P-distance的矩阵,表示两两物种间的分歧距离。MEGA就可以得到,多序列比对完有个计算。如下
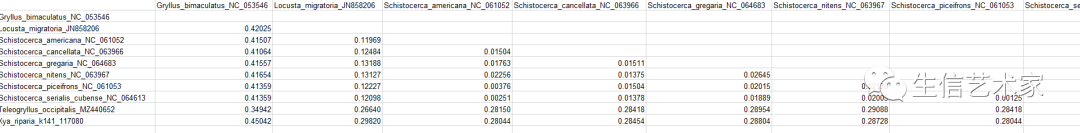
可以把这个p-distance矩阵用python读到数据框中
dt = pd.read_excel("pdistance.xls",sheet_name=[0],index_col=0)然后需要一个两两物种分歧时间的矩阵也读到数据框中
dt1 = pd.read_excel(input1,sheet_name=[0],index_col=0)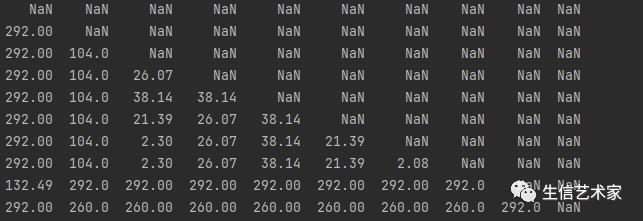
这个是两两物种分歧时间单位:百万年
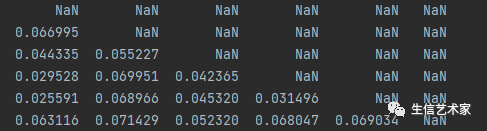
这个是两两物种间分歧距离也是两两物种间的碱基替换率。
接下来下写了个python脚本根据上面进化速率的公式计算即可
output_file = open(output, "w")k = 0f = 0allrate = 0for i in list:k = k + 1list1 = list[k:]for z in list1:linename = i + '_' + zMya =df.loc[z, i]if math.isnan(Mya):Mya = df.loc[i, z]Pdistance = df1.loc[z, i]if math.isnan(Pdistance):Pdistance = df1.loc[i, z]print( str(Mya) + '_' + str(Pdistance))rate = float(Pdistance) / (float(Mya)*2)output_file.write(linename + "\t" + str(rate) + '\n')f = f + 1allrate = allrate + ratemeanrate = float(allrate) / foutput_file.write('mean' + "\t" + str(meanrate) + '\n')output_file.close()
最后就会得到一个平均进化速率。
2.密码子同义替换与非同义替换速率
使用PAML 中baseml来计算
第一步安装
wget http://abacus.gene.ucl.ac.uk/software/paml4.9j.tgztar xf paml4.9j.tgzrm bin/*.execd srcmake -f Makefilerm *.omv baseml basemlg codeml pamp evolver yn00 chi2 ../bin
第二步 准备输入文件
paml的输入文件有三个,seq文件,tree文件,另外一个是配置文件,里面就是调整我们输入数据,输出数据,使用模型,进化速率等等的调整。
第三步 修改配置文件baseml.ctl
seqfile = stewart.aa * sequence data filenametreefile = stewart.trees * tree structure file nameoutfile = mlc * main result file namenoisy = 9 * 0,1,2,3,9: how much rubbish on the screenverbose = 1 * 0: concise; 1: detailed, 2: too muchrunmode = 0 * 0: user tree; 1: semi-automatic; 2: automatic* 3: StepwiseAddition; (4,5):PerturbationNNI; -2: pairwiseseqtype = 2 * 1:codons; 2:AAs; 3:codons-->AAsCodonFreq = 2 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:codon table* ndata = 10clock = 0 * 0:no clock, 1:clock; 2:local clock; 3:CombinedAnalysisaaDist = 0 * 0:equal, +:geometric; -:linear, 1-6:G1974,Miyata,c,p,v,aaaRatefile = dat/jones.dat * only used for aa seqs with model=empirical(_F)* dayhoff.dat, jones.dat, wag.dat, mtmam.dat, or your ownmodel = 2* models for codons:* 0:one, 1:b, 2:2 or more dN/dS ratios for branches* models for AAs or codon-translated AAs:* 0:poisson, 1:proportional, 2:Empirical, 3:Empirical+F* 6:FromCodon, 7:AAClasses, 8:REVaa_0, 9:REVaa(nr=189)NSsites = 0 * 0:one w;1:neutral;2:selection; 3:discrete;4:freqs;* 5:gamma;6:2gamma;7:beta;8:beta&w;9:betaγ* 10:beta&gamma+1; 11:beta&normal>1; 12:0&2normal>1;* 13:3normal>0icode = 0 * 0:universal code; 1:mammalian mt; 2-10:see belowMgene = 0* codon: 0:rates, 1:separate; 2:diff pi, 3:diff kapa, 4:all diff* AA: 0:rates, 1:separatefix_kappa = 0 * 1: kappa fixed, 0: kappa to be estimatedkappa = 2 * initial or fixed kappafix_omega = 0 * 1: omega or omega_1 fixed, 0: estimateomega = .4 * initial or fixed omega, for codons or codon-based AAsfix_alpha = 1 * 0: estimate gamma shape parameter; 1: fix it at alphaalpha = 0. * initial or fixed alpha, 0:infinity (constant rate)Malpha = 0 * different alphas for genesncatG = 8 * # of categories in dG of NSsites modelsgetSE = 0 * 0: don't want them, 1: want S.E.s of estimatesRateAncestor = 1 * (0,1,2): rates (alpha>0) or ancestral states (1 or 2)Small_Diff = .5e-6cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)?* fix_blength = 1 * 0: ignore, -1: random, 1: initial, 2: fixed, 3: proportionalmethod = 0 * Optimization method 0: simultaneous; 1: one branch a time* Genetic codes: 0:universal, 1:mammalian mt., 2:yeast mt., 3:mold mt.,* 4: invertebrate mt., 5: ciliate nuclear, 6: echinoderm mt.,* 7: euplotid mt., 8: alternative yeast nu. 9: ascidian mt.,* 10: blepharisma nu.* These codes correspond to transl_table 1 to 11 of GENEBANK.
seqfile = test.codon * sequence data filenametreefile = test.tree * tree structure file nameoutfile = test.rlt * main result file namenoisy = 0 * 0,1,2,3,9: how much rubbish on the screenverbose = 0 * 0: concise; 1: detailed, 2: too muchrunmode = -2 * 0: user tree; 1: semi-automatic; 2: automatic* 3: StepwiseAddition; (4,5):PerturbationNNI; -2: pairwise###此处模式选择一般是0或者-2,0表示使用你生成的tree,-2表示两两比较seqtype = 1 * 1:codons; 2:AAs; 3:codons-->AAsCodonFreq = 2 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:codon table* ndata = 5504 ###此处即为多少配对的数目,就我们上面统计的clock = 0 * 0:no clock, 1:clock; 2:local clock; 3:CombinedAnalysisaaDist = 0 * 0:equal, +:geometric; -:linear, 1-6:G1974,Miyata,c,p,v,aaaRatefile = dat/jones.dat * only used for aa seqs with model=empirical(_F)* dayhoff.dat, jones.dat, wag.dat, mtmam.dat, or your ownmodel = 0* models for codons:* 0:one, 1:b, 2:2 or more dN/dS ratios for branches* models for AAs or codon-translated AAs:* 0:poisson, 1:proportional, 2:Empirical, 3:Empirical+F* 6:FromCodon, 7:AAClasses, 8:REVaa_0, 9:REVaa(nr=189)NSsites = 0 * 0:one w;1:neutral;2:selection; 3:discrete;4:freqs;* 5:gamma;6:2gamma;7:beta;8:beta&w;9:betaγ* 10:beta&gamma+1; 11:beta&normal>1; 12:0&2normal>1;* 13:3normal>0icode = 0 * 0:universal code; 1:mammalian mt; 2-10:see belowMgene = 0* codon: 0:rates, 1:separate; 2:diff pi, 3:diff kapa, 4:all diff* AA: 0:rates, 1:separatefix_kappa = 0 * 1: kappa fixed, 0: kappa to be estimatedkappa = 2 * initial or fixed kappafix_omega = 0 * 1: omega or omega_1 fixed, 0: estimateomega = .4 * initial or fixed omega, for codons or codon-based AAsfix_alpha = 1 * 0: estimate gamma shape parameter; 1: fix it at alphaalpha = 0. * initial or fixed alpha, 0:infinity (constant rate)Malpha = 0 * different alphas for genesncatG = 8 * # of categories in dG of NSsites modelsgetSE = 0 * 0: don't want them, 1: want S.E.s of estimatesRateAncestor = 1 * (0,1,2): rates (alpha>0) or ancestral states (1 or 2)Small_Diff = .5e-6cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)?* fix_blength = 1 * 0: ignore, -1: random, 1: initial, 2: fixed, 3: proportionalmethod = 0 * Optimization method 0: simultaneous; 1: one branch a time* Genetic codes: 0:universal, 1:mammalian mt., 2:yeast mt., 3:mold mt.,* 4: invertebrate mt., 5: ciliate nuclear, 6: echinoderm mt.,* 7: euplotid mt., 8: alternative yeast nu. 9: ascidian mt.,* 10: blepharisma nu.* These codes correspond to transl_table 1 to 11 of GENEBANK.
可以参考上面这个配置文件,修改好了之后就可以运行baseml
###配置完成后,开始运行baseml baseml.ctl
结果后就可以得到一下结果文件
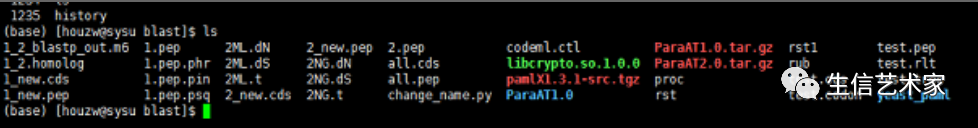
其中包含dN,dS,rst这些为结果文件,根据需要去画图。
以上就是物种分歧时间、分子进化速率的所有内容了,包括了两种方法推断物种分歧时间(mcmctree 和 MEGA reltime),TimeTree查询化石节点,计算分子进化速率(核酸替换速率、密码子同义替换速率与非同义替换速率)。
如果感觉有帮助的话,下面来个硬币吧!

参考链接
-
http://www.chenlianfu.com/?p=2974
-
https://www.jianshu.com/p/f9e5fe95478d
-
http://www.fish-evol.org/mcmctreeExampleVert6/text1Eng.html
-
http://abacus.gene.ucl.ac.uk/software/MCMCtreeStepByStepManual.pdf
-
https://dosreislab.github.io/2017/10/24/marginal-likelihood-mcmc3r.html
-
http://nebc.nerc.ac.uk/bioinformatics/documentation/paml/doc/MCMCtreeDoc.pdf
https://www.jianshu.com/p/b12e058c6597
https://zhuanlan.zhihu.com/p/105159767
https://www.sci666.com.cn/20394.html
以上内容仅为个人总结,如有错误之处敬请批评!
联系我们:
生信艺术家

















 4409
4409

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









