






1. 先下载SRR数据
ascp -QT -l 10m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR137/094/SRR13755394/SRR13755394.fastq.gz .......其他的类似自行下载
2. 数据过滤及质控
fastqc SRR13755394.fastq.gz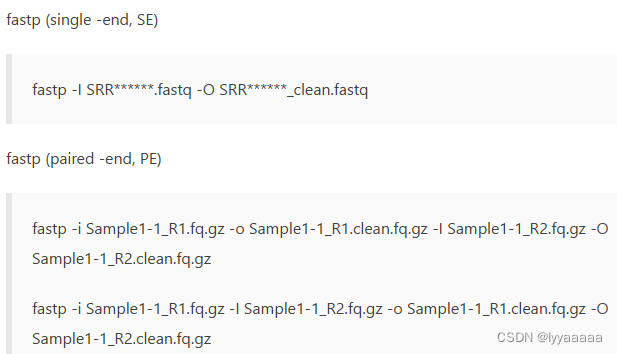
3. 准备参考基因组
cd /student2/data/RNAseq/raw/fq
mkdir references
wget https://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/hg19.fa.gz![]()
4. 索引参考基因组(选用STAR)
STAR --runMode genomeGenerate --runThreadN 2 --genomeDir star --genomeFastaFiles ./references/hg19.fa --sjdbGTFfile ./references/gencode.v19.annotation.gtf
5. 创建map.sh比对
先解压gunzip SRR13755394.fastq.gz(其他类似)
for file in 'SRR13755394' 'SRR13755395' 'SRR13755396' 'SRR13755397' 'SRR13755398' 'SRR13755399' 'SRR13755400' 'SRR13755401' 'SRR13755402' 'SRR13755403' 'SRR13755404' 'SRR13755405'
do
echo $file
STAR --quantMode GeneCounts --runThreadN 2 --genomeDir star --readFilesIn $file.fastq --outFileNamePrefix ./align/$file --outFilterMultimapNmax 1 --outSAMtype BAM SortedByCoordinate
Done
6. 计数count.sh
for file in *ReadsPerGene.out.tab; do
basename="${file%ReadsPerGene.out.tab}"
cut -f1,4,8,12,16 "$file" | tail -n +5 > "${basename}.tmpfile"
cat "${basename}.tmpfile" | sed 's/^gene://' > "${basename}_gene_count.txt"
done
7. 合并merge.sh
#!/bin/bash
# 初始化输出文件
output_file="gene_expression_matrix.txt"
tmp_output="tmp_gene_expression.txt"
# 获取所有 *_gene_count.txt 文件的列表
files=(*_gene_count.txt)
# 提取所有基因的ID并写入gene_ids.txt(假设每个gene_count.txt文件的第一列是GeneID)
cut -f1 "${files[0]}" > gene_ids.txt
# 初始化临时文件并写入基因ID
cp gene_ids.txt "$tmp_output"
# 遍历所有 *_gene_count.txt 文件,合并到临时文件中
for file in "${files[@]}"; do
sample_name="${file%_gene_count.txt}"
echo "Processing $sample_name"
cut -f2 "$file" > "${sample_name}_counts.tmp"
paste "$tmp_output" "${sample_name}_counts.tmp" > tmp && mv tmp "$tmp_output"
done
# 添加表头
header="GeneID"
for file in "${files[@]}"; do
sample_name="${file%_gene_count.txt}"
header="$header\t$sample_name"
done
sed -i "1s/^/$header\n/" "$tmp_output"
# 将结果文件重命名为最终输出文件
mv "$tmp_output" "$output_file"
















 429
429











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









