






1. 首先下载SRR数据(这里举一个例子,其他的类似)
ascp -QT -l 10m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR137/037/SRR13755437/SRR13755437.fastq.gz .
ascp -QT -l 10m -P33001 -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR155/099/SRR15567099/SRR15567099.fastq.gz .2. 使用fastqc做质量检查
3. 解压
gunzip SRR13755437.fastq.gz
gunzip SRR15567099.fastq.gz4. bowite2建立索引
bowtie2-build hg19.fa hg195. bowite2比对(这里批量比对)
新建一个脚本
cd ./data/chip-seq/raw
touch bowtie.sh
脚本内容
#!/bin/bash
REFERENCE="/student2/data/chip-seq/references/hg19"
FASTQ_FILES=("SRR13755437.fastq" "SRR13755438.fastq" "SRR15567099.fastq" "SRR15567100.fastq")
for FASTQ in "${FASTQ_FILES[@]}"
do
SAMPLE_NAME=$(basename "$FASTQ" .fastq)
#打印当前处理的文件
echo "Processing $SAMPLE_NAME"
bowtie2 -p 5 -t -x $REFERENCE -U $FASTQ | samtools sort -O bam -o ./${SAMPLE_NAME}.bam
#检查命令是否成功执行
if [ $? -eq 0 ]; then
echo "$SAMPLE_NAME processing completed successfully."
else
echo "Error processing $SAMPLE_NAME."
fi
done
echo "ALL samples processed."
6. 构建index文件
为了在igv里查看peak,需要把bam文件先构建index,即生成bam.bai文件:
samtools index SRR13755437.bam SRR13755437.bam.bai
samtools index SRR15567099.bam SRR15567099.bam.bai
7. 用deeptools里的bamcovera工具将bam文件标准化
bamCoverage -b SRR13755437.bam -o SRR13755437.bw -p 10 --normalizeUsing RPKM
bamCoverage -b SRR15567099.bam -o SRR15567099.bw -p 10 --normalizeUsing RPKM将生成的bw文件(标准化后的导入)导入IGV
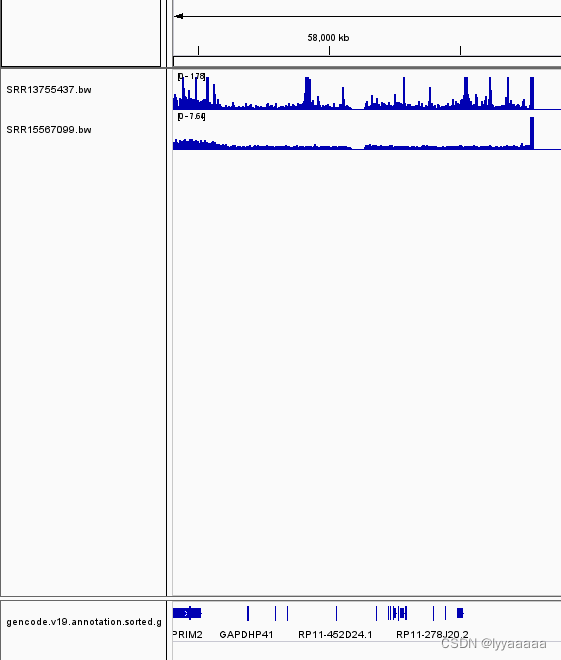
8. 使用macs3获得chip-seq富集区
macs3 callpeak -c SRR15567099.bam -t SRR13755437.bam -m 10 30 -p 1e-5 -f BAM -g hs -n HMEC_rep1 --call-summits-c:对照组
-t:处理组
--format:输入的文件类型
--:输出的文件前缀名称
关于宽峰和窄峰:
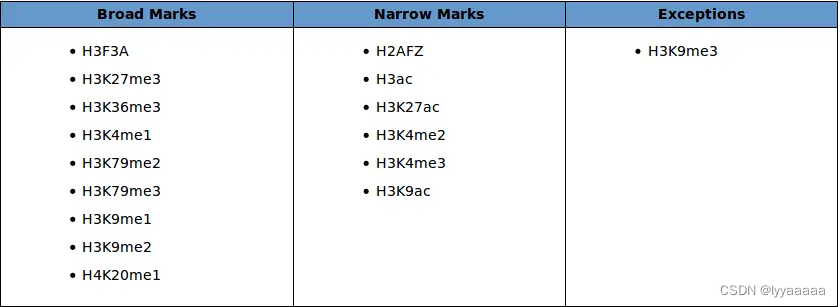
生成:
9. ROSE筛选super-enhancer
(1)第一步先安装rose(可以参考下面的链接)
(2)安装后,准备文件:peaks的gff文件,这个peak是MACS3 call peak 之后生成的peaks文件

第1列:染色体位置(chr#)
第2列:每个增强子区域的特定id
第4列:区域起始位置
第5列:区域终止位置
第7列:正负链信息(+, -, .)
第9列:每个增强子区域的特定id
上面没有要求的列,内容可以为空或者原来的内容,但是一定要有这列,如果第2列和第9列的内容不同,ROSE将使用第2列的值。
awk '{print $1"\t"$4"\t"" ""\t"$2"\t"$3"\t"" ""\t"$6"\t"" ""\t"$4}' HMEC_rep1_peaks.narrowPeak > HMEC_rep1_peaks3.gffpython ./bin/ROSE_main.py -g HG19 -i ./data/HMEC_rep1_peaks3.gff -r ./data/SRR13755437.bam -c ./data/SRR15567099.bam -o roseresult -s 12500 -t 2500运行过程中如果报以下错误:
Traceback (most recent call last):
File "/hpcstor6/scratch01/m/mingyu.liu001/Super_Enhancer/ROSE-1.3.0/bin/ROSE_geneMapper.py", line 291, in
main()
File "/hpcstor6/scratch01/m/mingyu.liu001/Super_Enhancer/ROSE-1.3.0/bin/ROSE_geneMapper.py", line 277, in main
enhancerToGeneTable,geneToEnhancerTable = mapEnhancerToGene(annotFile,enhancerFile,uniqueGenes=True,byRefseq=options.refseq, transcribedFile=transcribedFile)
File "/hpcstor6/scratch01/m/mingyu.liu001/Super_Enhancer/ROSE-1.3.0/bin/ROSE_geneMapper.py", line 145, in mapEnhancerToGene
closestGene = allEnhancerGenes[distList.index(min(distList))]
IndexError: list index out of range
这是因为在生成的txt结果文件中又不规范的染色体名比如chr1_sgdy类似等等,修改ROSE_main.py加入:
def filter_non_standard_chromosomes(file_path):
# 读取文件内容
with open(file_path, 'r') as f:
lines = f.readlines()
# 找到有效数据起始行
start_idx = 0
for idx, line in enumerate(lines):
if line.startswith('REGION_ID'):
start_idx = idx
break
# 处理有效数据部分
processed_lines = []
for line in lines[start_idx:]:
if line.startswith('#') or line.strip() == '':
processed_lines.append(line) # 保留注释和空行
else:
# 按制表符分割行数据
parts = line.split('\t')
# 获取原始的染色体名
original_chrom = parts[1].strip()
# 判断染色体名是否符合标准形式
if original_chrom.startswith('chr') and (
original_chrom[3:].isdigit() or original_chrom[3:] in ['X', 'Y']):
# 符合标准形式,保留
processed_lines.append('\t'.join(parts))
# 更新原始文件内容
with open(file_path, 'w') as f:
# 先写入无效信息部分
for line in lines[:start_idx]:
f.write(line)
# 再写入处理后的有效数据部分
for line in processed_lines:
f.write(line)
print(f"文件 {file_path} 已成功更新,不符合标准染色体的行已删除。")
superTableFile = "%s/%s_SuperStitched.table.txt" % (outFolder,inputName)
allTableFile = "%s/%s_AllStitched.table.txt" % (outFolder,inputName)
filter_non_standard_chromosomes(superTableFile)
filter_non_standard_chromosomes(allTableFile)结果文件:
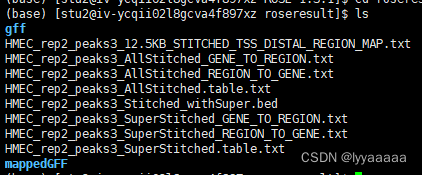
怎么看结果可以参考我下面的链接
10. ROSE注释enhancer
python ./bin/ROSE_geneMapper.py -i ./roseresult/HMEC_rep2_peaks3_AllStitched.table.txt -g HG19 -o ./roseresult 2> ./roseresult/enhancer_anno.log
运行结束生成以下结果:
HMEC_rep2_peaks3_AllStitched_GENE_TO_REGION.txt
HMEC_rep2_peaks3_AllStitched_REGION_TO_GENE.txt
具体怎么看参考以下链接
参考链接:
ChIP-seq实践(H3K27Ac,enhancer的筛选和enhancer相关基因的GO分析) - 简书 (jianshu.com)
Super Enhancer(超级增强子)分析——ROSE包(v1.3.1)的安装及使用详解_超级增强子rose-CSDN博客

















 7996
7996

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









