







文章目录
前言
本文是古生物谱系建树相关内容分享。
注:不论是古生物形态学数据或DNA数据,就建树方法来说是一样的,所以本文中也用DNA数据的例子。
一、相关背景
1.1 数据类型(本文介绍常见的两种)
形态学数据:

通过观察化石图片,古生物草图等得到,有某一特征则标记为1,没有记为0。如上图,物种A有尾巴记作1,没尾巴记作0。
DNA数据:

可以提取到DNA的物种便可以得到DNA数据。
以上两个矩阵叫做特征矩阵。
DNA数据构建系统发育树更为精确,那为什么要研究形态学数据呢?
可想而知古生物是难以提取DNA的,所以只能通过形态学数据来建树。
1.2 建树流程
先大致明白系统发育树构建流程,如果数据完整(特征矩阵完整)我们通过一些算法直接就构建系统发育树啦。

举例(距离矩阵法):

二、数据问题
数据问题主要是两个方面。
2.1数据集缺失
由于化石的损坏,某些特征缺失了。缺失数据标记为“?”。

解决方案:
删除缺失数据:某个物种缺失过多,或某个特征缺失过多,那就将其删除。
增加特征数量:通过不断发现新化石来增添特征。
模糊优化:将缺失变为一个已经有的状态或者新得状态。
插补:数据插补是一个常见的对缺失数据的操作,但是在古生物方面这或许不是一个好的选择,我们不能保证插补的正确,插补错误造成的后果难以衡量。
忽略:缺失数据不做什么处理,只通过存在的数据建树。
2.2 数据不可适用问题
由于建树时化石物种之间相对差异较大,存在特征缺失,即数据不可适用。
举例说明:现在搜集特征尾巴的颜色(红黄蓝等),而物种A的尾巴不存在,所以根本谈不上是哪个颜色。只能记作 “-”,即为不可适用。

解决方案:
还原编码:用“?” 代替 “-”。
复合编码:将特征简化,避免出 “-”。
……
常见的处理不可适用数据的方式都是对不可适用数据的代替或者规避,没有从不可适用数据中挖掘信息。
推荐这样一篇文章:An algorithm for Morphological Phylogenetic Analysis with Inapplicable Data (不可适用Fitch法建树)
三、建树方法介绍
3.1简约法
先知道一个必须要明白的概念。
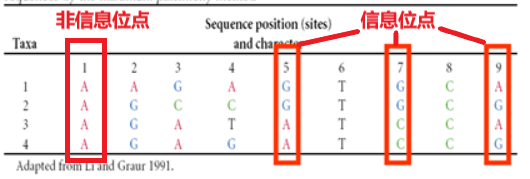
信息位点:能将所有可能的树区别出来的位点。
如下图中的位点1,都是碱基A,那在这个位点是得不到信息的,像5,7,9这样的信息位点才能提供给我们信息。
最大简约法(Maximum Parsimony )是系统发生分析中最简单最经典的方法之一。
这种方法的理论前提是,越少的变化是越可能的。举例:对于人的进化,我们更倾向于从有尾巴到没尾巴,而不是从没尾巴到有尾巴到没尾巴。

上图中,不同的颜色代表不同的信息位点,具体来说,考虑seeds(图中蓝色)这个特征(位点),上图中左侧拓扑结构pine tree(叶节点)是1,父节点是0,从0到1发生变化要标记,同理可得树其他位置的标记,也可得其他位点的标记,所有的标记数相加就得到了这棵树的树长,树长越小的树越可能是真的树。
上图中左侧树树长=7,右侧树树长=3,那我们认为右侧的树更为正确。
3.2似然法
最大似然法原理:将系统树的拓扑结构、分支长度、进化模型参数等 的全部或部分作为需要估计的参数
θ
\theta
θ
,在给定数据集和进化模型的基础上,用最大似然法的标准–似然值最大化来估计这些参数。
关键的三点:
进化模型参数:可在模型选择的过程中就确定,或者通过最大似然法估计。
分支长度(速率*时间):将其与似然值对数作图,似然值最大时对应的值就是分支长度的最大似然估计值。如下图

拓扑结构:目前的ML法,我们不是通过最大似然法来估计一个拓扑结构(可想而知拓扑结构不能用确切的数值表示,当然不能放在似然函数里估计出来),而是在“有好的分支长度的估计值的拓扑结构可能是真实树”这一假设下,选择一个具有最大ML值的拓扑结构。简单来说,在某个拓扑下进化模型、分支长度已经估计出来,计算得到似然值,比较不同拓扑的似然值,似然值更大的树是我们想要的树。
最大似然法建树流程:
(1)选择进化模型
(2)以MPT或NJ树(距离法)为基础采样ML法估计模型中的各个参数
(3)从MP(最大简约)树、NJ树或其他的起始树开始进行ML分析,以发现似然值更大的树。
(4)用统计学方法从多个ML树中选择最优ML树。
eg1: 最大似然法 (2)在系统发育的具体应用.
eg2: 这是一个非常好的例子!明白这个例子就明白了最大似然法建树过程!


3.3贝叶斯法
P ( T r e e ∣ D a t a ) = P ( D a t a ∣ T r e e ) P ( T r e e ) P ( D a t a ) P(Tree|Data)=\frac{P(Data|Tree)P(Tree)}{P(Data)} P(Tree∣Data)=P(Data)P(Data∣Tree)P(Tree)
我们可以看出式子右侧有什么,先验值和似然值。
下图很好的展示了贝叶斯法的过程。
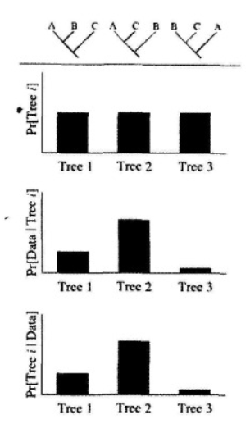
后验概率越大的树越是我们想要的,然而贝叶斯这种方法遇到了计算上的困难。
后验概率不仅涉及所有可能的树,而且对于每一棵树还考虑分支长度和替换模型参数值的所有可能组合,所以不可能采用常规方法计算。所幸MCMC(马尔可夫蒙特卡洛)算法可模拟近似获得后验概率。
MCMC通过采集样本来近似树的后验分布,具体来说就是从初始树出发,通过分枝交换操作(见后文)得到下一个树,这个树再通过分枝交换操作,得到下一个树……通过这样运行得到大量的树样本来近似树的后验分布。一个树出现的频率就是树的后验概率值,贝叶斯法建树就是寻找后验概率值最大的树,往往有很多个树的后验概率值一样大,那么把这些树进行合意操作,得到一个最终的合意树。
MCMC的详细介绍请前往本人的另一篇博客
注:3.2及3.3的内容很多参考了《分子系统发生学》这本书.
四、树如何进行最优化搜索
这部分内容讲的是什么呢,因为通过前面的建树方法来看,随着物种数,特征数增多,我们不可能把每一个可能的拓扑列出来一一计算,我们只能从某些树(比如距离法构建了一颗树作为初始树)出发,不断变化树的结构得到新得树,这个过程就需要用到树的分支交换操作。
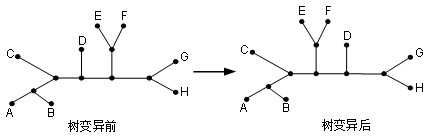
4.1最近邻交换(NNI)
内部分支一侧的分支与另一个分支交换位置。
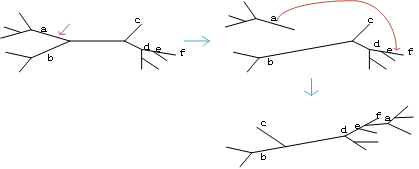
4.2子树修剪和嫁接(SPR)
将剪下来的部分插到剩余树的终端分支或者内部分支上。
4.3树分割和重连(TBR)
将一个树拆成两个子树,然后将这两个子树以任意方式重连。
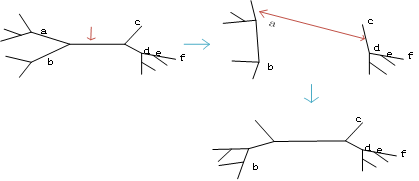
以上三种方式其实TBR包含SPR,SPR包含NNI.
TBR是最广泛使用的方法,但当分类单元增多时,其不能保证搜索到最优树。(可能陷入到局部最优了)
五、系统发育树度量指标
找出来的树如何评判好坏,有以下几个常用的指标。
5.1树长
不同特征在系统发育树中的特征状态变换次数的总和,这个值越小越好。
5.2一致性指数
C
I
=
m
s
CI =\frac{m}{s}
CI=sm
m为最小可能的树长,s表示当前系统发育树的树长,这个值接近1越好。
5.3RF距离
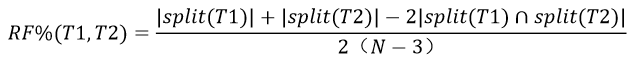
可以计算树之间的距离,值越小表明距离越近。
最后:
笔者并不是生物专业,文中错误之处欢迎指出。
某些图片忘记截取自哪里了,如有侵权请多海涵。

















 506
506

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









