






前言
经常需要画简单的系统发育树(不用知道基因序列),但每次要画的时候都忘记前一次的辛酸学习过程,因此决定记录下来。本文没有创新内容,参考全部来自其他网站,只是做了简单的归纳总结。
一、前期准备
已知的物种名、物种的性状值(将性状值和系统发育树合并)、R语言plantlist包(具体可以参考科学网—用plantlist程序包查询和处理植物学名 (plantlist 0.6.5) - 张金龙的博文 (sciencenet.cn))、ape包
二、步骤
1、将物种名在plantlist中查询,得到科属种结果
假设我要生成以下物种的系统发育树:蜡梅(Chimonanthus praecox)、含笑花(Michelia figo)、苏铁(Cycas revoluta)、银杏(Ginkgo biloba)、豺皮樟(Litsea rotundifolia)、木棉(Bombax ceiba)、凤凰木(Delonix regia)、榕树(Ficus microcarpa)、杧果(Mangifera indica)、龙眼(Dimocarpuslongan)、乌桕(Triadica sebifera)、枫香树(Liquidambar formosana)、糖胶树(Alstonia scholaris)、樟(Camphora officinarum)
library(plantlist)
taxa<-c("Chimonanthus praecox",
"Michelia figo",
"Cycas revoluta",
"Ginkgo biloba",
"Litsea rotundifolia",
"Bombax ceiba",
"Delonix regia",
"Ficus microcarpa",
"Mangifera indica",
"Dimocarpus longan",
"Triadica sebifera",
"Liquidambar formosana",
"Alstonia scholaris",
"Camphora officinarum")
TAXA<-TPL(taxa)
TAXA
#输出结果如下:(取前五行)
#YOUR_SEARCH POSSIBLE_GENUS FAMILY FAMILY_NUMBER ORDER GROUP
#1 Chimonanthus praecox Chimonanthus Calycanthaceae APGIII_022 Laurales #Angiosperms
#2 Michelia figo Michelia Magnoliaceae APGIII_017 Magnoliales #Angiosperms
#3 Cycas revoluta Cycas Cycadaceae Gymnosperm_01 Cycadales #Gymnosperms
#4 Ginkgo biloba Ginkgo Ginkgoaceae Gymnosperm_03 Ginkgoales #Gymnosperms
#5 Litsea rotundifolia Litsea Lauraceae APGIII_028 Laurales #Angiosperms
taxa.table(TAXA)#将查询结果转为“科/属/种”格式
#输出结果如下:(取前五行)
#[1] "Calycanthaceae/Chimonanthus/Chimonanthus_praecox"
#[2] "Magnoliaceae/Michelia/Michelia_figo"
#[3] "Cycadaceae/Cycas/Cycas_revoluta"
#[4] "Ginkgoaceae/Ginkgo/Ginkgo_biloba"
#[5] "Lauraceae/Litsea/Litsea_rotundifolia"
P.S. 若拉丁名为excel的一列,从excel导入:
taxa<-read_excel("data.xlsx")
taxa1<-as.vector(unlist(taxa))
taxa2<-TPL(taxa1)
TAXA<-taxa.table(taxa2)
#注:excel中物种间的空在R中可能为<U+00A0>,此时需要在excel中替换为空格将结果写入txt文件write.table(TAXA,file="...",col.names=FALSE,row.names=FALSE,quote=FALSE)2、根据得到的科/属/种结果画括号形式的进化树
首先需要安装rtools或git,将awk.exe添加到系统路径中,同时下载phylomatic-awk-main.zip(GitHub - camwebb/phylomatic-awk: A CLI phylomatic written in Gawk)
此处参考(科学网—在本地运行awk版的phylomatic利用植物名录生成进化树 - 张金龙的博文 (sciencenet.cn))
将phylomatic-awk-main.zip解压缩,打开文件夹右击选择git bash here,在命令行窗口输入以下命令:
gawk -f phylomatic --clean --newick data/zanne2014.new --taxa examples/TAXA
会得到如下结果:
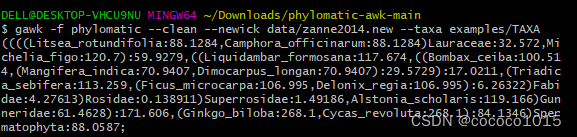
注❗:此处将第一步中的TAXA文件复制到phylomatic-awk-main中的examples文件夹中,将TAXA的后缀“.txt”删去,即可运行以上代码。
保存此步生成的括号形式的进化树:
gawk -f phylomatic --clean --newick data/zanne2014.new --taxa examples/TAXA>tree.txt此时在phylomatic-awk-main文件夹中会出现“tree.txt"的文件
3、画图
ape包的read.tree()函数可以读取txt格式的树文件,画图代码如下:
plot(read.tree("tree.txt"))生成简单的进化树:
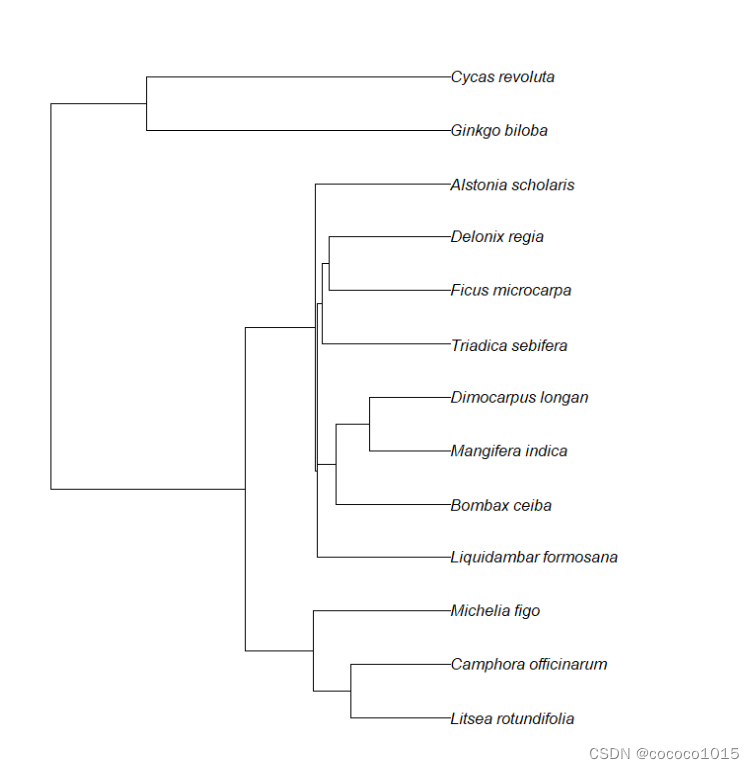
图片美化可以继续参考学习:DrawingPhylogenies.pdf (rdrr.io)
P.S. read.tree()函数也可以读取".new"格式的文件。
phylomatic的相关网站:
参考博文里还提到的V.PhyloMaker包和rtrees包画树,尚未深入学习,学习后再进行更新。
补充:更加方便的画树方式参考(如何画物种进化树? - 知乎)
三、将进化树和性状值相结合
看到很多文献经常画这样子的图,简单学习了一下,但还不知道可以怎样解释这种图,以下粗略总结了过程。(具体参考的文献有点不记得了,这里就没有列出)
接下来用另外14个物种进行举例
1、准备括号形式的进化树
根据"二、步骤----->2"生成的“tree.txt"
2、准备数据
df<-read_excel(...)#文件名、路径根据具体情况而定
df<-na.omit(df)
head(df)
#以下为输出结果A tibble: 6 x 4 species biomass length RD <chr> <dbl> <dbl> <dbl> 1 Liquidambar formosana 0.196 1272. 0.259 2 Liquidambar formosana 0.128 517. 0.297 3 Liquidambar formosana 0.135 560. 0.315 4 Liquidambar formosana 0.113 1235. 0.276 5 Liquidambar formosana 0.252 1412. 0.262 6 Liquidambar formosana 0.0887 372. 0.276
注:数据第一列为物种名,此处前六行数据均为一个物种
#求各数据的均值
bio<-aggregate(df$biomass,by=list(type=df$species),mean)
len<-aggregate(df$length,by=list(type=df$species),mean)
rd<-aggregate(df$RD,by=list(type=df$species),mean)
species<-gsub(" ","_",bio$type)#因为画图需要,将物种的空格改为下划线
data<-data.frame(species,bio$x,len$x,rd$x)#合并所有列
colnames(data)<-c("","biomass","length","RD")#为每一列命名
rownames(data)<-data[,1]#将第一列作为行名
data<-data[,-1]#删除第一列数据data已经可以用于画图了。
注:这里注意这段代码的最后两行,要将第一列作为行名,生成的“data”在R中展示如下:
head(data)
# biomass length RD
#Carpinus_turczaninowii 0.05259861 599.2311 0.1364543
#Chamaecyparis_obtusa 0.12255636 420.6921 0.3013540
#Cryptomeria_japonica 0.08038349 232.1351 0.4245821
#Cunninghamia_lanceolata 0.28883708 417.3095 0.5617870
#Cyclocarya_paliurus 0.10557623 933.1948 0.1770841
#Eurya_muricata 0.04320539 600.7784 0.1408129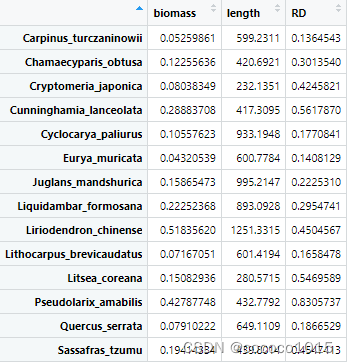
3、画图
画图需要用到phylobase包的phylo4d()函数和phylosignal包的barplot.phylo4d()函数,代码如下:
p4d <- phylo4d(read.tree("tree.txt"), data)
barplot.phylo4d(p4d, tree.type = "phylo", tree.ladderize = TRUE)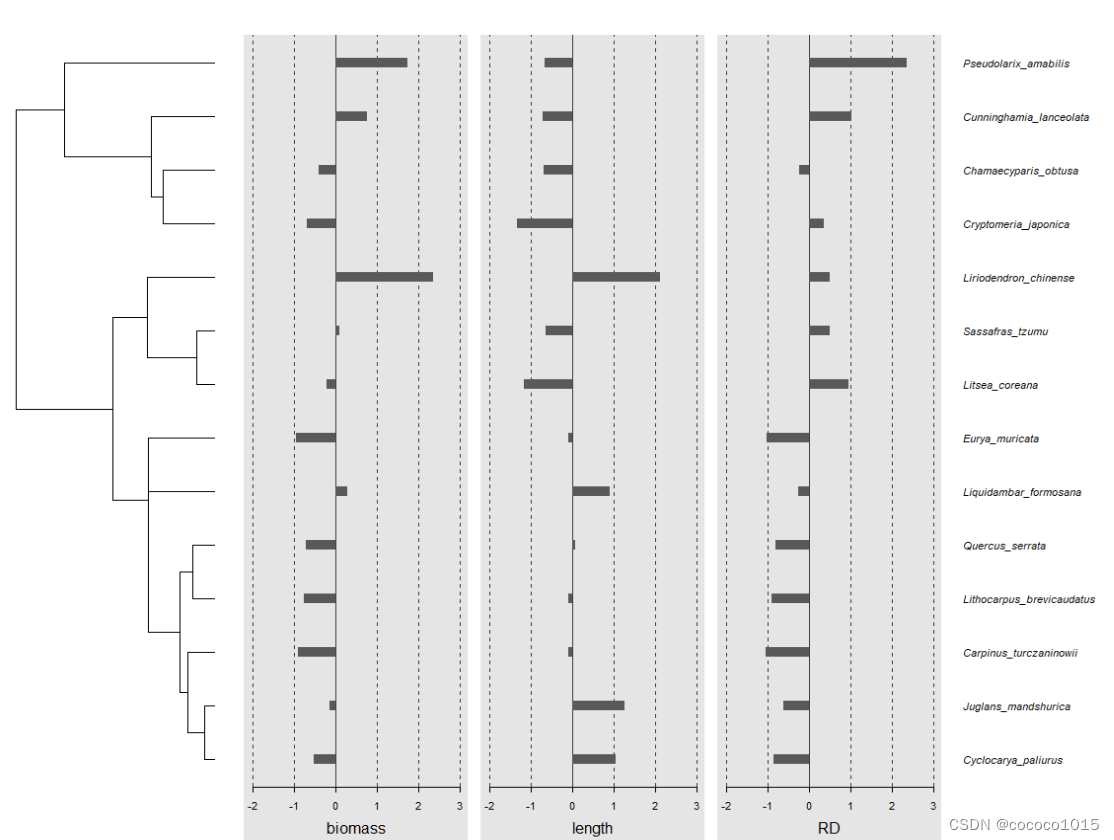
图片美化可以学习以上R包的详细介绍。
phylosignal包相关文献:phylosignal: an R package to measure, test, and explore the phylogenetic signal - Keck - 2016 - Ecology and Evolution - Wiley Online Library
备注:此处仅用平均值进行计算,好像还可以计算其他一些指标;只试过植物,关于其他物种的画图可以参考文献或R包;可能还有其他方式画,还没有找到相关资料,待更新。
四、计算PIC(系统发育独立差)
参考来源:科学网—什么是系统发育独立差(Phylogenetic Independent Contrast)? - 张金龙的博文 (sciencenet.cn)
整理成如下格式的数据:(R语言中的展示)
head(data)
# species biomass length RD
#1 Carpinus_turczaninowii 0.05259861 599.2311 0.1364543
#2 Chamaecyparis_obtusa 0.12255636 420.6921 0.3013540
#3 Cryptomeria_japonica 0.08038349 232.1351 0.4245821
#4 Cunninghamia_lanceolata 0.28883708 417.3095 0.5617870
#5 Cyclocarya_paliurus 0.10557623 933.1948 0.1770841
#6 Eurya_muricata 0.04320539 600.7784 0.1408129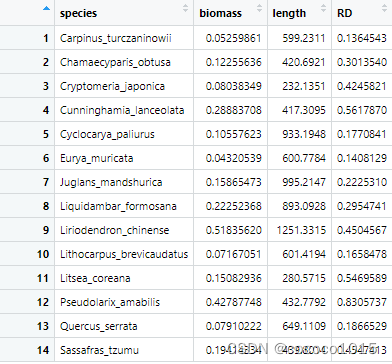
注:此处与“三、将进化树和性状值相结合------>2、准备数据”中最后生成的样式不同,此处的物种名仍为单独一列。
以下为参考博文中的代码流程及一些解释说明:
biomass<-data$biomass
names(biomass)<-data$species #这里使biomass的数据形式变为“named num”,可以用于后续的匹配
species<-gsub("_"," ",as.character(data$species))#这里要把物种中的下划线"_"再改为空格
#如果数据中本身就是空格此步可以忽略
#以下几行代码实际上是用之前提到的V.PhyloMaker包画进化树,生成“.new”(newick)格式的文件
taxa<-subset(TPL(species),select=c("YOUR_SEARCH","POSSIBLE_GENUS","FAMILY"))
colnames(taxa)<-c("species","genus","family")
result1<-phylo.maker(taxa,scenarios=c("S1"))
#write.tree(result1[[1]],"tree.newick")
#ape的pic函数不能处理含有多分枝结构的进化树,所以用multi2di()函数,将进化树转换为二叉树
tree<-multi2di(result1[[1]])
#match.phylo.data函数能将性状数据按照进化树中物种出现的顺序排好
data_matched <- match.phylo.data(phy = tree, data = biomass)
#计算PIC
PIC<- pic(data_matched$data, data_matched$phy)结果如下:
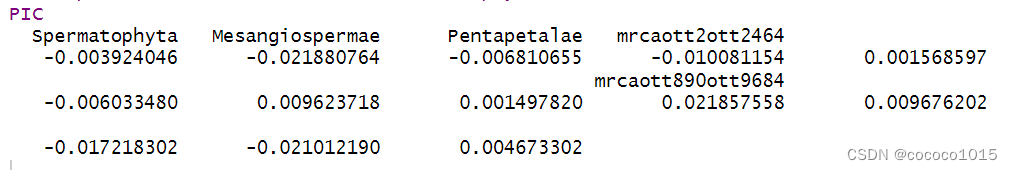
(该括号内内容不用看
稍微改动一点,可以和之前生成过的数据衔接上的代码流程:
biomass<-data$biomass
names(biomass)<-data$species
tree<-multi2di(read.tree("tree.txt"))
data_matched <- match.phylo.data(phy = tree, data = biomass)
PIC<- pic(data_matched$data, data_matched$phy)
#length和RD也是一样地写代码计算结果如下:

注:下面代码和第一次代码生成结果不同,个人认为是因为不同R包生成进化树时物种的上下顺序不同,但应该不影响结果。
更新:以上括号内内容似乎不可行,但是暂时还未发现原因,先保留着。大家可以绕过不看。)
若要得到计算PIC后的相关系数,就直接利用cor()函数即可。
五、最后
其实里面很多内容都参考了张金龙博士张老师的博客,从开始学习画树到现在,帮助真的特别大,特别感谢老师!

















 540
540

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









