










DPA-1: 共建覆盖元素周期表的预训练大模型 - 知乎 (zhihu.com)
深度势能学习(1)—基本知识 - 知乎 (zhihu.com)
深度势能学习(2)—DeepMD-kit学习 - 知乎 (zhihu.com)
(2 封私信) 有哪些值得推荐的分子动力学模拟入门书籍? - 知乎 (zhihu.com)
RuNNer Documentation (theochemgoettingen.gitlab.io)
RuNNer - Georg-August-Universität Göttingen (uni-goettingen.de)
#下面这个知乎讲的非常好!!!
Transformer模型详解(图解最完整版) - 知乎 (zhihu.com)
Seq2Seq模型介绍 - 知乎 (zhihu.com)
end-to-end 模式比较:
DeepPot-SE || DeePMD [17] || GDML [11] || SchNet [12]
end-to-end(DeepPot-SE)
[[报告/报告7_有限和扩展系统的端到端对称性保持原子间势能模型|报告7_有限和扩展系统的端到端对称性保持原子间势能模型]]
本文 模型比较:
DPA-1 (OC2M -pretrained) || DPA-1 || EANN || DeePot-SE
EANN:
[[报告9_EANN]]
Yaolong Zhang, Ce Hu, and Bin Jiang. Embedded atom neural network potentials: Efficient and accurate machine learning with a physically inspired representation. The journal of physical chemistry letters, 10(17):4962–4967, 2019.
我们提出了一个简单但高效且准确的机器学习(ML)模型,用于开发高维势能表面。这种[所谓的嵌入式原子神经网络 (EANN) 方法的灵感来自凝聚相中使用的众所周知的经验嵌入式原子方法 (EAM) 模型。它只是用基于高斯型轨道的密度向量替换了EAM中的标量嵌入原子密度,并通过神经网络表示嵌入密度向量与原子能之间的复杂关系。]我们证明,EANN方法在表示大分子和扩展周期系统方面与几个已建立的ML模型一样准确,但参数和配置要少得多。它非常高效,因为它隐式包含三体信息,而没有传统昂贵的角度描述符的显式总和。凭借高精度和高效率,EANN 电位可以极大地加速复杂系统中的分子动力学和光谱模拟。
这句话描述了嵌入式原子神经网络 (EANN) 方法的灵感和基本思想,这是一种用于分子模拟的机器学习潜力。 EANN 方法的灵感来自嵌入原子方法 (EAM),这是一种广泛使用的用于模拟金属和合金行为的经验潜力。EAM势通过将能量分解为成对相互作用和多体嵌入能量来模拟系统的总能量,作为原子位置的函数。嵌入能量表示将原子嵌入其相邻原子的电子密度所需的能量,并表示为标量嵌入原子密度的函数。 EANN 方法通过将标量嵌入原子密度替换为密度函数向量来扩展 EAM 势,密度函数表示为以每个原子为中心的高斯型径向基函数。密度函数描述了每个原子周围的局部电子密度,并用作神经网络的输入,该神经网络预测系统的嵌入能量和总能量。 换句话说,EANN方法使用神经网络来学习密度函数和嵌入能量之间的复杂关系,而不是像EAM势那样使用预定义的函数形式。密度函数提供了更准确、更灵活的局部电子密度表示,神经网络可以捕获非线性和高阶效应,这对于预测总能量和原子力很重要。 EANN 方法已被证明可以准确有效地模拟各种材料,包括金属、合金和分子。它可用于大规模分子动力学模拟,有助于了解材料的微观结构演变、相变和力学行为。
下面翻译文献:《DPA-1: Pretraining of Attention-based Deep Potential Model for Molecular Simulation》
摘要
原子间势能面(PES)的机器学习辅助建模正在彻底改变分子模拟领域。随着高质量电子结构数据的积累,一个可以根据所有可用数据进行预训练并在下游任务中进行微调的模型将把该领域带入一个新阶段。在这里,我们提出了DPA-1,一个具有新的注意机制的深度势模型,它对于表示原子系统的构象和化学空间以及学习PES非常有效。我们在多个系统上测试了DPA-1,并观察到与现有基准相比具有更高的性能。当在包含56个元素的大规模数据集上进行预训练时,DPA-1可以成功地应用于各种下游任务,并大大提高了采样效率。令人惊讶的是,对于不同的元素,学习的类型嵌入参数在潜在空间中形成螺旋,并与它们在元素周期表中的位置具有自然对应关系,显示了预训练的DPA-1模型的有趣的可解释性。
1 介绍
在计算物理、化学、材料科学、生物学等领域,可靠地表示原子间势能面(PES)是研究分子和材料性质的核心。虽然电子结构方法通常给出精确和可转移的PES,但对于扩展到超过数千个原子的系统来说,它们非常昂贵。另一方面,经验力场更有效,但在许多应用中受到其准确性的固有限制。通过适当地集成机器学习(ML)方法和物理要求,如可扩展性和对称性,出现了各种方法来解决准确性问题。PES建模领域的效率困境【1–11】。可以说,一个新的范式正在形成:[电子结构方法]不再用于在分子动力学模拟期间产生驱动力,而是用于生成数据以训练其替代方案,即基于ML的PES模型。
*[电子结构方法]:
在分子动力学 (MD) 模拟中,传统上使用电子结构方法通过计算系统的势能面 (PES) 来产生驱动力。PES将系统的势能描述为核坐标的函数,并用于计算作用在原子核上的力,这决定了模拟中原子的运动。
电子结构法是一种量子力学方法,用于计算系统的电子结构,其中包括电子的能量和波函数。在MD模拟的背景下,电子结构方法用于计算系统的能量作为核坐标的函数。这是通过求解电子自由度的薛定谔方程来完成的,受制于原子核固定在一组给定位置的约束。对于原子核的许多不同配置重复此过程以生成 PES。
PES 通常表示为多维函数,每个核自由度都有一个维度。PES 上任何给定点的势能可用于计算作用在原子核上的力,这些力由势能相对于核坐标的负梯度给出。然后,根据运动方程,这些力用于更新原子核在模拟中的位置。
近年来,人们越来越关注使用机器学习 (ML) 方法生成用于 MD 模拟的 PES。这些基于ML的PES模型在电子结构计算数据集上进行训练,它们能够准确预测原子核新构型的势能和力。使用基于ML的PES模型有可能显著降低MD模拟的计算成本,因为它们不需要对原子核的每个新配置求解薛定谔方程。
总体而言,电子结构方法一直是在 MD 模拟中产生驱动力的重要工具,但它正越来越多地被基于 ML 的 PES 模型所取代,这些模型提供了一种更有效、更准确的方法来计算这些模拟中的势能和力。
尽管基于ML的PES模型取得了显著的成就【12–14】,挑战仍然存在。对于想在应用程序中应用这些方法的领域专家来说,一个自然的首要问题是获得一个可靠的PES模型所需的努力:是否有现成的PES模型?—如果不是,需要多少训练数据量和时间成本?我们能利用不断增加的公开培训数据吗?
为了解决这些问题,已经做出了一些努力。一方面,各种系统的通用模型,如硅[15]、磷[16]、水[17]、金属和合金【18-22】等。,已经开发并直接适用于相关研究。然而,这种模型的适用范围通常局限于小构象或化学空间。例如,对于合金,大多数通用ML模型是为最多具有两种元素类型的系统开发的。另一方面,已经开发了几种有效的[数据生成协议]【23–26】,其中一个代表是DP-GEN[25,26],这是一种并发学习过程,它使用用现有数据训练的模型迭代探索配置空间,然后仅标记那些具有高不确定性水平的配置。即使有了这些协议,复杂系统所需的计算工作量仍然令人望而却步。例如,为了为AlMgCu合金系统训练一个相当通用的模型,最终进行了100k密度泛函理论(DFT)[27,28]计算,导致了一千万CPU核心小时的成本[18]。
*[数据生成协议]:
数据生成协议是用于生成配置和相应属性数据集的方法,可用于训练用于分子模拟的机器学习 (ML) 模型。这些协议对于确保 ML 模型在多样化且具有代表性的数据集上进行训练非常重要,这对于它们准确预测感兴趣的属性是必要的。
数据生成协议的一个例子是 DP-GEN,它是一个并发学习过程,它使用在现有数据上训练的模型来迭代探索配置空间并识别具有高度不确定性的配置。这是通过首先在配置和相应属性的小型数据集上训练 ML 模型来完成的。然后,该模型用于预测一组较大配置的属性,并选择具有最高不确定性的配置进行进一步研究。然后,通过电子结构计算或其他方式使用这些配置来生成新数据,并使用这些新信息更新数据集。
重复迭代探索配置空间和识别高不确定性配置的过程,直到收集到足够数量的数据点。然后使用最终数据集来训练新的ML模型,由于数据的多样性和代表性增加,预计该模型将比初始模型更准确和可靠。
DP-GEN是一种强大的数据生成协议,因为它允许有效地探索配置空间和识别高不确定性配置,这对于准确建模感兴趣的属性通常是最重要的。通过使用基于现有数据训练的模型来指导探索,DP-GEN能够专注于配置空间中最相关的区域,这有助于降低生成数据集的计算成本。
总体而言,像 DP-GEN 这样的数据生成协议是训练用于分子模拟的准确可靠的 ML 模型的重要工具。通过仔细设计数据生成过程,可以确保 ML 模型在多样化且具有代表性的数据集上进行训练,这对于它们准确预测感兴趣的属性是必要的。
随着涵盖元素周期表上几乎所有元素的高质量电子结构数据的积累,系统地开发预训练方案变得可能,这些方案已被广泛应用于计算机视觉(CV)[29,30]和自然语言处理(NLP)[31,32]等领域。在这些方案中,首先在大规模数据集上训练统一的模型,然后针对下游任务对其进行微调,期望在第一阶段可以学习到良好的表示,并且第二阶段所需的监督数据量将显著减少。不幸的是,大多数基于ML的PES模型对于大规模的这种方案来说是不成熟的。以广泛使用的两个版本的深度势模型[6,7]为例,ML参数是元素类型相关的,这使得当训练数据包含许多元素时效率非常低。尽管最近有一些开发**“通用”PES模型**的尝试[33,34],但它们在下游任务和大规模应用上的微调性能还有待观察。
在本文中,我们提出了DPA-1,这是一种具有新型注意机制的深度电位模型[35,30],它对于学习原子间的相互作用非常有效,并且在预训练后,可以显著减少下游任务的额外努力。
我们在各种系统上测试了DPA-1,并观察到与现有基准相比的卓越性能。然后我们以AlMgCu合金系统[18]为例,表明在用单元素和二元样本进行预训练后,与DeepPot-SE模型[7]相比,DPA-1可以节省大约90%的三元样本。
最后,我们使用Open Catalyst 2020数据集(OC20)[36]对DPA-1进行了预训练,该数据集由56个元素组成,并成功地将其应用于各种下游任务。我们通过查看不同元素类型的学习嵌入参数来检查预训练模型的可解释性,发现56种元素在潜在空间中排列成螺旋,这与它们在元素周期表中的物理性质具有自然对应关系。最重要的是,我们相信DPA-1和预训练方案将把分子模拟领域带到一个新的阶段。
本文提出了Al-Cu-Mg合金在全浓缩空间下的精确深势模型。
深度势模型 是一种机器学习势能,可以高精度地预测材料系统的势能和原子力。该模型基于深度势分子动力学 (DPMD) 框架,该框架使用人工神经网络从从头开始计算中学习材料系统的势能面。
作者使用具有不同成分和结构的Al-Cu-Mg合金的从头计算的大型数据集训练了深势模型。该数据集包括10,000多个结构和2000万个原子,覆盖了Al-Cu-Mg合金的全浓度空间。深势模型可以高精度地预测Al-Cu-Mg合金的势能和原子力,与从头计算的精度相当。 作者通过将预测的特性与实验数据和从头开始的计算进行比较,验证了深度电位模型。结果表明,深势模型能够准确预测Al-Cu-Mg合金的结构、力学和热力学性质,包括晶格常数、弹性常数、声子谱和地层能等。 深势模型可用于Al-Cu-Mg合金的大规模分子动力学模拟,有助于理解合金的微观结构演化、相变和力学行为。深势模型还可用于合金成分和加工参数的设计和优化。 综上所述,本文提出了Al-Cu-Mg合金在全浓缩空间下的精确深势模型,有利于Al-Cu-Mg合金的计算材料科学研究。通过考虑更多的训练数据、更复杂的神经网络架构和更先进的机器学习技术,可以进一步改进该模型。
2 方法
考虑一个由N个原子组成的系统,元素类型是
A
\mathcal A
A={
α
1
,
α
2
,
.
.
.
,
α
i
,
.
.
.
,
α
N
α_1,α_2,...,α_i,...,α_N
α1,α2,...,αi,...,αN},原子坐标是
R
\mathcal R
R={
r
1
,
r
2
,
.
.
.
,
r
i
,
.
.
.
,
r
N
r_1,r_2,...,r_i,...,r_N
r1,r2,...,ri,...,rN},其中
r
i
r_i
ri是原子i的三个笛卡尔坐标。系统的PES用E表示,E是元素类型和坐标的函数,即E=E(
A
\mathcal A
A,R)。对于每个原子i,考虑它的邻居{j | j∈
N
r
c
\mathcal N_{r_c}
Nrc(i)},其中
N
r
c
\mathcal N_{r_c}
Nrc(i)表示原子指数j的集合,使得
r
j
i
<
r
c
r_{ji}<r_c
rji<rc,其中
r
j
i
r_{ji}
rji是原子i和j之间的欧几里德距离。
E表示为原子能的总和{
e
1
,
e
2
,
.
.
.
,
e
i
,
.
.
.
,
e
N
e_1,e_2,...,e_i,...,e_N
e1,e2,...,ei,...,eN},其中原子能
e
i
e_i
ei只取决于
N
r
c
\mathcal N_{r_c}
Nrc(i)的信息。我们定义
N
i
N_i
Ni=
N
r
c
\mathcal N_{r_c}
Nrc(i),集合
N
r
c
\mathcal N_{r_c}
Nrc(i)的基数。我们用
A
i
\mathcal A^i
Ai表示
N
r
c
\mathcal N_{r_c}
Nrc(i)中的元素类型,
R
i
R^i
Ri∈
R
N
i
×
3
R^{N_i × 3}
RNi×3表示它们相对于i的对应坐标。因此,原子能
e
i
e_i
ei是
A
i
\mathcal A^i
Ai和
R
i
R^i
Ri的函数。原子i上的原子力
F
i
F_i
Fi定义为总能量相对于i坐标的负梯度:
F
i
=
−
∇
r
i
E
.
(1)
\mathcal{F}_i=-\nabla_{\boldsymbol{r}_i}E.\tag{1}
Fi=−∇riE.(1)
![![[Pasted image 20240414145303.png]]](https://i-blog.csdnimg.cn/blog_migrate/987f7eb92e860d1c5a0a438ae6c2ec16.png)
图1:DPA-1的示意图。
(a)从
A
i
A^i
Ai和
R
i
R^i
Ri到原子能
e
i
e_i
ei的流程图。
(b)嵌入网的结构,其通过多个残差层将
s
(
r
j
i
)
s(r_{ji})
s(rji)和
T
i
T_i
Ti映射到
G
i
\mathcal G^i
Gi。
©通过由角度信息门控的标准标度-点程序在
G
i
\mathcal G^i
Gi上的自我注意机制。
(d)拟合网络结构,类似于嵌入网络,从描述符
D
i
D^i
Di和
T
i
T_i
Ti到最终原子能
e
i
e_i
ei。
关于PES建模的几个要求的详细讨论见[[报告/报告7_有限和扩展系统的端到端对称性保持原子间势能模型|报告7_有限和扩展系统的端到端对称性保持原子间势能模型]],请参考文献[7]。特别是,在具有相同元素类型的原子的指数的平移、旋转和排列下,PES必须是不变的。下面介绍模型架构的细节。我们参考图1来预测原子能ei的整个流水线:从嵌入的邻近环境,通过自我注意方案,到对称保持描述符,最后到拟合网络。
1 具有类型信息的局部嵌入矩阵
我们通过以下三个步骤获得局部嵌入矩阵。首先,
R
i
R^i
Ri被映射到广义坐标
R
~
i
\tilde{\mathcal{R}}^{i}
R~i∈
R
N
i
×
4
R^{N_i × 4}
RNi×4。在这个映射中,
R
i
R^i
Ri,{
x
j
i
,
y
j
i
,
z
j
i
x_{ji},y_{ji},z_{ji}
xji,yji,zji}的每一行都被转换成Ri的一行:
{
x
j
i
,
y
j
i
,
z
j
i
}
↦
{
s
(
r
j
i
)
,
x
^
j
i
,
y
^
j
i
,
z
^
j
i
}
,
(2)
\{x_{ji},y_{ji},z_{ji}\}\mapsto\{s\left(r_{ji}\right),\hat{x}_{ji},\hat{y}_{ji},\hat{z}_{ji}\},\tag{2}
{xji,yji,zji}↦{s(rji),x^ji,y^ji,z^ji},(2)
{ x j i , y j i , z j i } 表示的笛卡尔坐标 r j i = r j − r i , x ^ j i = s ( r j i ) x j i r j i , y ^ j i = s ( r j i ) y j i r j i , z ^ j i = s ( r j i ) z j i r j i \mathrm\{x_{ji},y_{ji},z_{ji}\}\text{表示的笛卡尔坐标}r_{\boldsymbol{ji}}=r_{\boldsymbol{j}}-r_{\boldsymbol{i}},\hat{x}_{ji}=\frac{s(r_{ji})x_{ji}}{r_{ji}},\hat{y}_{ji}=\\\frac{s(r_{ji})y_{ji}}{r_{ji}},\hat{z}_{ji}=\frac{s(r_{ji})z_{ji}}{r_{ji}} {xji,yji,zji}表示的笛卡尔坐标rji=rj−ri,x^ji=rjis(rji)xji,y^ji=rjis(rji)yji,z^ji=rjis(rji)zji
s(rji):R→R是应用于每个分量的连续可微标量加权函数,定义为:
s
(
r
j
i
)
=
{
1
r
j
i
r
j
i
<
r
c
s
1
r
j
i
[
u
3
(
−
6
u
2
+
15
u
−
10
)
+
1
]
r
c
s
≤
r
j
i
<
r
c
,
u
=
r
j
i
−
r
c
s
r
c
−
r
c
s
.
0
r
c
≤
r
j
i
(3)
s\left(r_{ji}\right)=\begin{cases}\frac{1}{r_{ji}}&r_{ji}<r_{cs}\\\frac{1}{r_{ji}}\left[u^3\left(-6u^2+15u-10\right)+1\right]&r_{cs}\leq r_{ji}<r_c,\quad u=\frac{r_{ji}-r_{cs}}{r_c-r_{cs}}.\\0&r_c\leq r_{ji}\end{cases}\tag{3}
s(rji)=⎩
⎨
⎧rji1rji1[u3(−6u2+15u−10)+1]0rji<rcsrcs≤rji<rc,u=rc−rcsrji−rcs.rc≤rji(3)
平滑截止参数的作用是使函数在截止半径内逐渐变为零,以实现平滑衰减的效果。具体而言,在截止半径 r c r_c rc 内,函数的值会逐渐减小,直到最终达到零。这种平滑的衰减过程可以使函数在局部区域的边界处更加平滑,避免出现突变或不连续性。
其次,我们添加原子类型嵌入作为补充信息。对于原子i,类型嵌入映射
T
i
T_i
Ti定义为:
T
i
=
ϕ
T
(
α
i
)
(4)
T_i=\phi_T(\alpha_i)\tag{4}
Ti=ϕT(αi)(4)
其中α i是原子i的原子类型,
ϕ
T
\phi_T
ϕT是从α i到长度固定向量的类单热嵌入网络映射。
定义了局部嵌入矩阵
G
i
∈
R
N
i
×
M
1
\mathcal{G}^i\in\mathbb{R}^{N_i\times M_1}
Gi∈RNi×M1:
(
G
i
)
j
=
G
(
s
(
r
j
i
)
,
T
i
,
T
j
)
,
(5)
\\\left(\mathcal{G}^i\right)_j=G\left(s\left(r_{ji}\right),T_i,T_j\right),\tag{5}
(Gi)j=G(s(rji),Ti,Tj),(5)
其中G是从标量权重 s ( r j i ) s(r_{ji}) s(rji)和中心原子和相邻原子的类型嵌入,通过多个隐藏层,到 M 1 M_1 M1输出的神经网络映射。这里,我们简单地将串联的输入一次馈入G,如图1(b)所示。
2 2.2建立可训练描述符的注意方法
注意机制取得了巨大的成功,并在CV[42]和NLP[43]中发挥了越来越重要的作用。它已经成为建模视觉区域或文本标记的重要性或相关性的优秀工具,因此潜在地适合于根据距离和角度信息重新加权相邻原子之间的相互作用。
这里采用Transformer model,完全基于注意力机制,完全免除了递归和卷积。
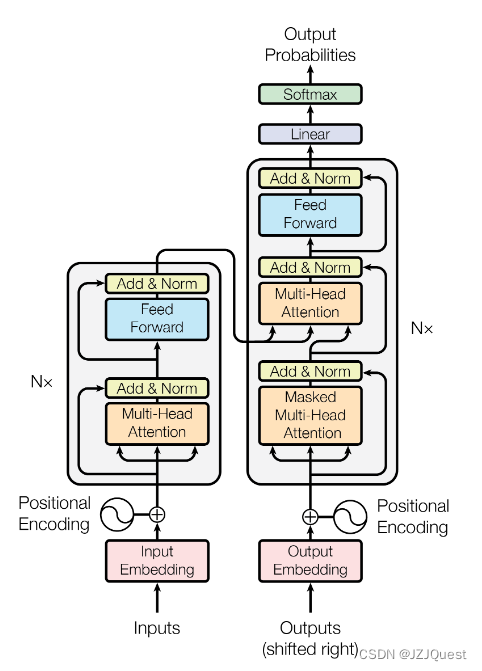
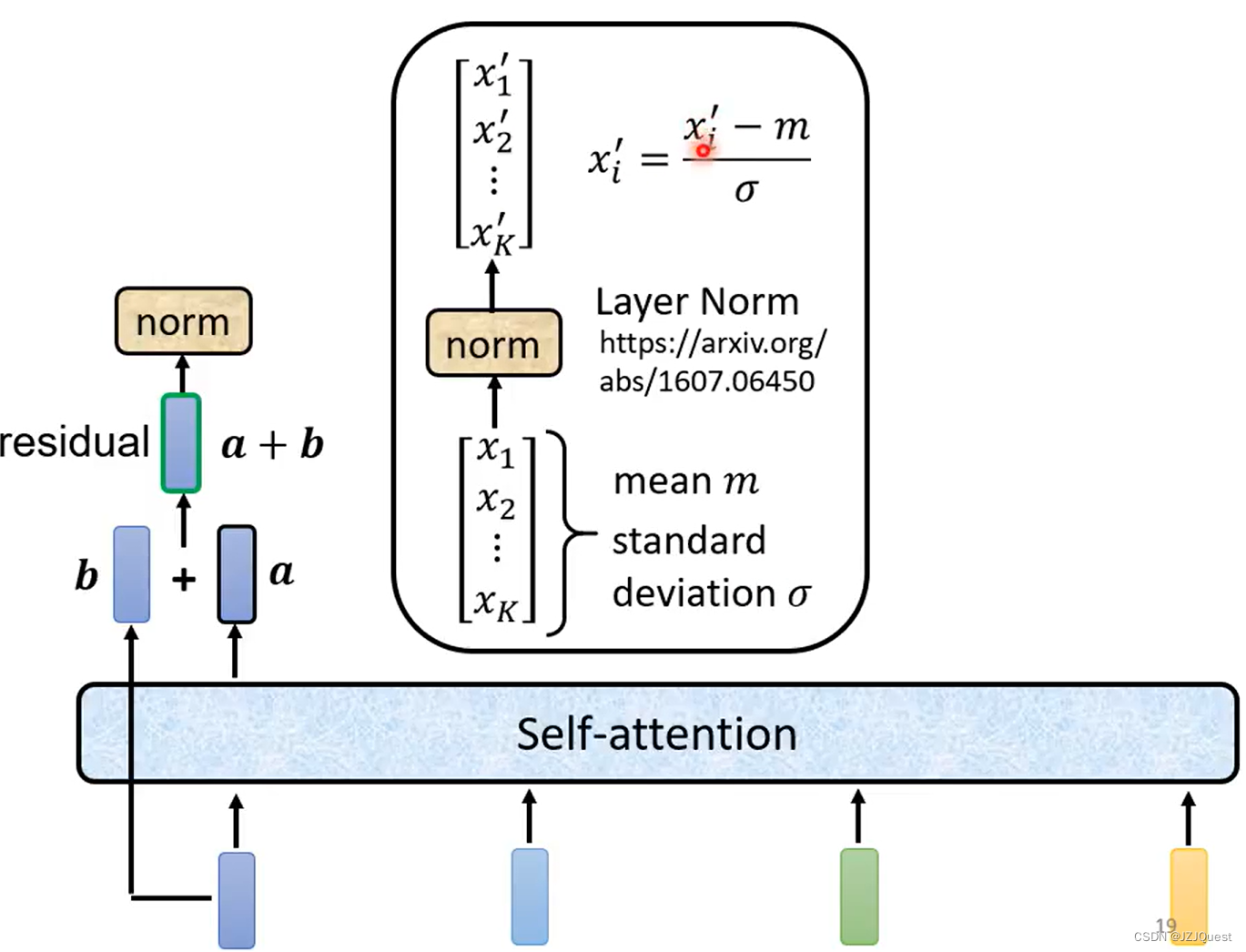
下面解释一下Add & Norm层的原理:
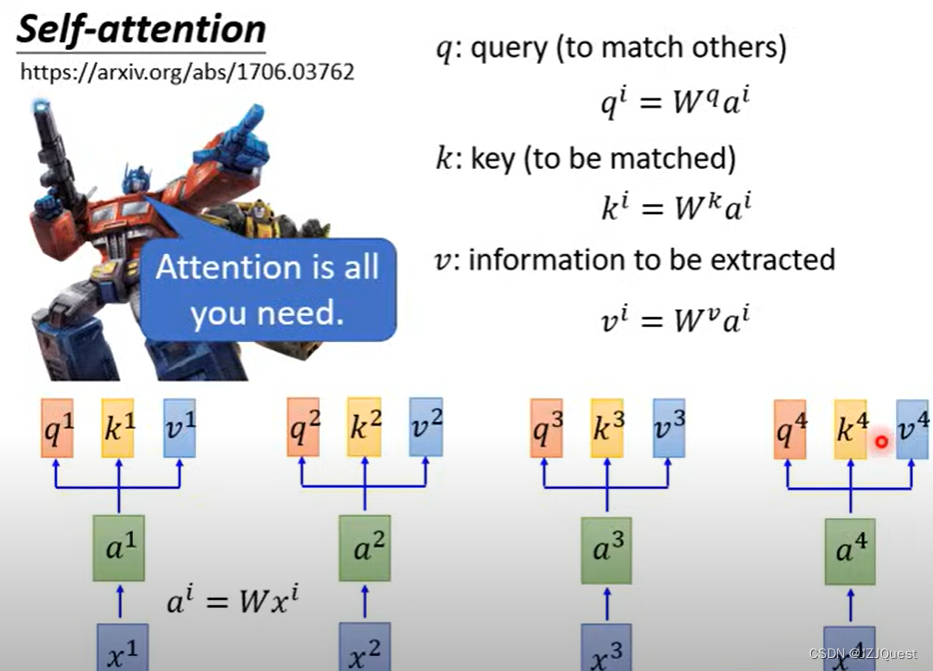
我们遵循标准的自我注意机制,得到查询
Q
i
,
l
∈
R
N
i
×
d
k
\mathcal{Q}^{i,l}\in\mathbb{R}^{N_i\times d_k}
Qi,l∈RNi×dk,关键字
K
i
,
l
∈
R
N
i
×
d
k
\mathcal{K}^{i,l}\in\mathbb{R}^{N_i\times d_k}
Ki,l∈RNi×dk,以及值
Q
i
,
l
∈
R
N
i
×
d
k
\mathcal{Q}^{i,l}\in\mathbb{R}^{N_i\times d_k}
Qi,l∈RNi×dk:
(
Q
i
,
l
)
j
=
Q
l
(
(
G
i
,
l
−
1
)
j
)
,
(
K
i
,
l
)
j
=
K
l
(
(
G
i
,
l
−
1
)
j
)
,
(
V
i
,
l
)
j
=
V
l
(
(
G
i
,
l
−
1
)
j
)
(6)
\left(\mathcal{Q}^{i,l}\right)_j=Q_l\left(\left(\mathcal{G}^{i,l-1}\right)_j\right),\left(\mathcal{K}^{i,l}\right)_j=K_l\left(\left(\mathcal{G}^{i,l-1}\right)_j\right),\left(\mathcal{V}^{i,l}\right)_j=V_l\left(\left(\mathcal{G}^{i,l-1}\right)_j\right) \tag{6}
(Qi,l)j=Ql((Gi,l−1)j),(Ki,l)j=Kl((Gi,l−1)j),(Vi,l)j=Vl((Gi,l−1)j)(6)
在这里,“标准的自我注意力机制”(standard self-attention mechanism) 通常指的是 Transformer 模型中的自我注意力机制(self-attention mechanism)。自我注意力机制允许模型在序列数据(例如,一段文本)中学习依赖关系,即哪些部分彼此更相关。
在Transformer模型中,自我注意力机制被分解为三个步骤:查询(Query)、关键字(Key)和值(Value)。这三个步骤使用相同的输入序列,每个步骤都有一个权重矩阵( Q l , K l , V l Q_l, K_l, V_l Ql,Kl,Vl) 和一个可学习的偏置( b l Q , b l K , b l V b_l^Q, b_l^K, b_l^V blQ,blK,blV)。
给定输入序列 G i , l − 1 ∈ R N i × d m o d e l \mathcal{G}^{i,l-1} \in \mathbb{R}^{N_i \times d_{model}} Gi,l−1∈RNi×dmodel,其中 N i N_i Ni 是序列长度, d m o d e l d_{model} dmodel 是输入维度。查询、关键字和值通过以下公式计算:
( Q i , l ) j = Q l ( ( G i , l − 1 ) j ) + b l Q \left(\mathcal{Q}^{i,l}\right)_j=Q_l\left(\left(\mathcal{G}^{i,l-1}\right)_j\right)+b_l^Q (Qi,l)j=Ql((Gi,l−1)j)+blQ
( K i , l ) j = K l ( ( G i , l − 1 ) j ) + b l K \left(\mathcal{K}^{i,l}\right)_j=K_l\left(\left(\mathcal{G}^{i,l-1}\right)_j\right)+b_l^K (Ki,l)j=Kl((Gi,l−1)j)+blK
( V i , l ) j = V l ( ( G i , l − 1 ) j ) + b l V \left(\mathcal{V}^{i,l}\right)_j=V_l\left(\left(\mathcal{G}^{i,l-1}\right)_j\right)+b_l^V (Vi,l)j=Vl((Gi,l−1)j)+blV
其中 j j j 是序列中的位置索引, Q l , K l , V l Q_l, K_l, V_l Ql,Kl,Vl 是权重矩阵, b l Q , b l K , b l V b_l^Q, b_l^K, b_l^V blQ,blK,blV 是可学习的偏置。
在计算注意力得分之前,计算出查询、关键字和值后,需要将它们分别 normalize 到同一空间,通常使用 scaled dot-product 来计算注意力得分:
A t t e n t i o n ( Q i , l , K i , l , V i , l ) = s o f t m a x ( Q i , l T K i , l d k ) V i , l Attention\left(\mathcal{Q}^{i,l},\mathcal{K}^{i,l},\mathcal{V}^{i,l}\right)=softmax\left(\frac{{\mathcal{Q}^{i,l}}^T\mathcal{K}^{i,l}}{\sqrt{d_k}}\right)\mathcal{V}^{i,l} Attention(Qi,l,Ki,l,Vi,l)=softmax(dkQi,lTKi,l)Vi,l
这里, d k d_k dk 是查询和关键字的维度,softmax 函数确保注意力得分的和为 1,从而保证概率的合法性。注意力得分表示了输入序列中每个位置与其他位置的相关性。
最终,自我注意力机制会输出一个新的序列 G i , l \mathcal{G}^{i,l} Gi,l,其中每个位置的值是基于整个序列计算出的注意力得分的加权和。
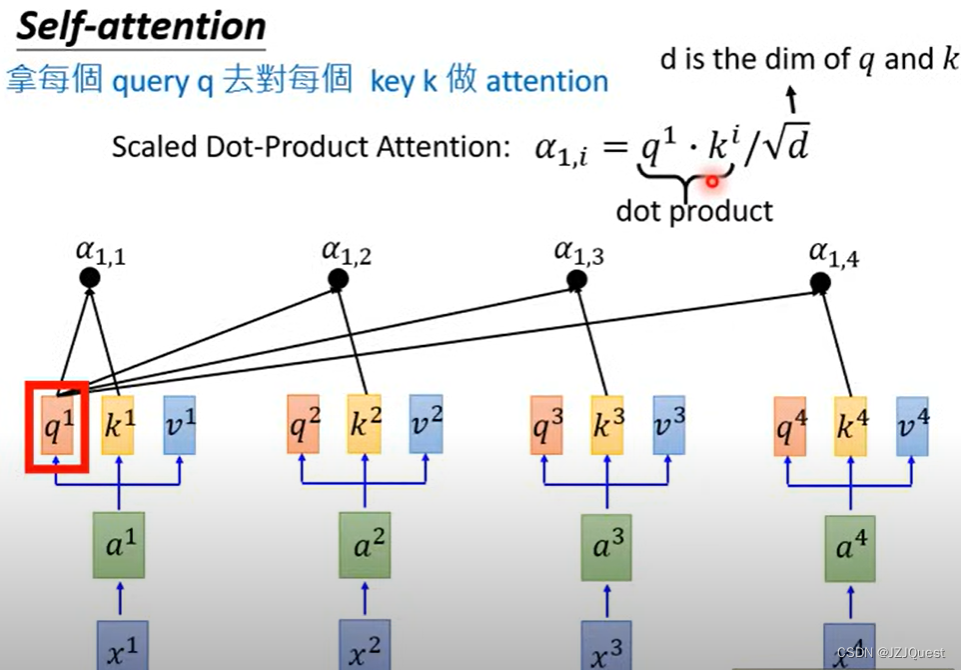
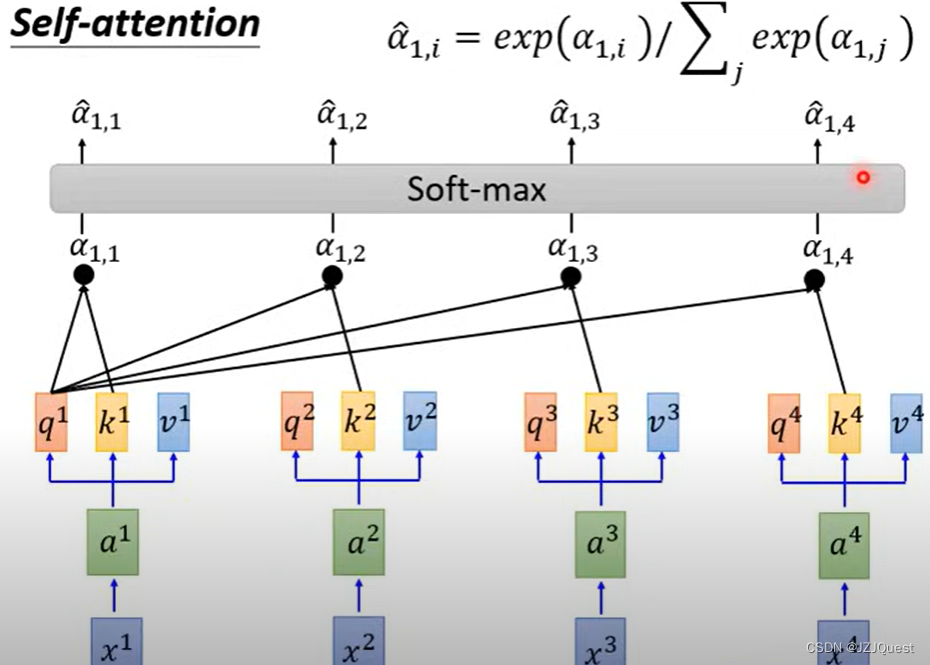
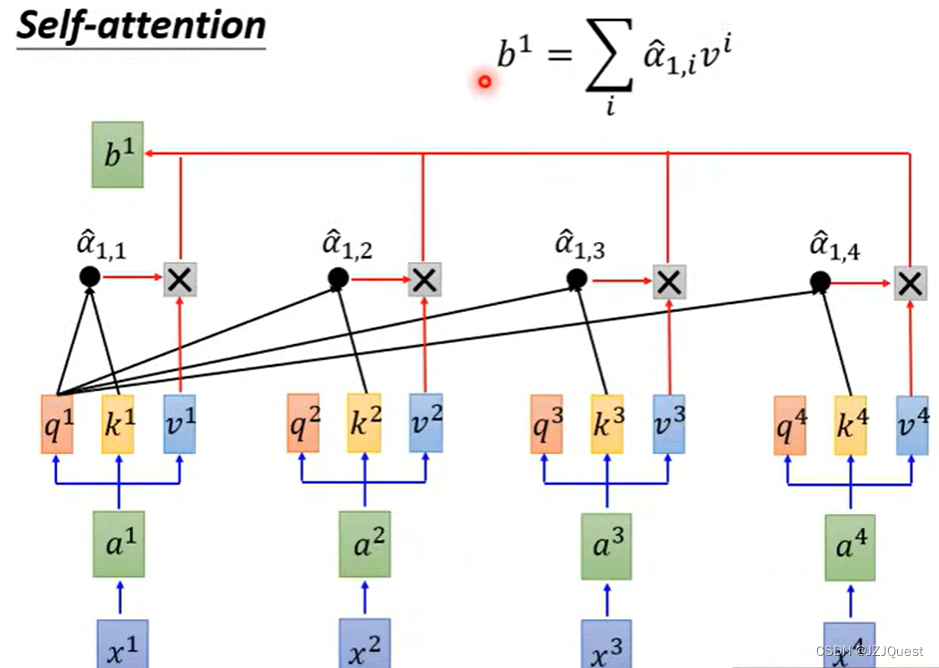
同理,
b
2
b^2
b2计算如下: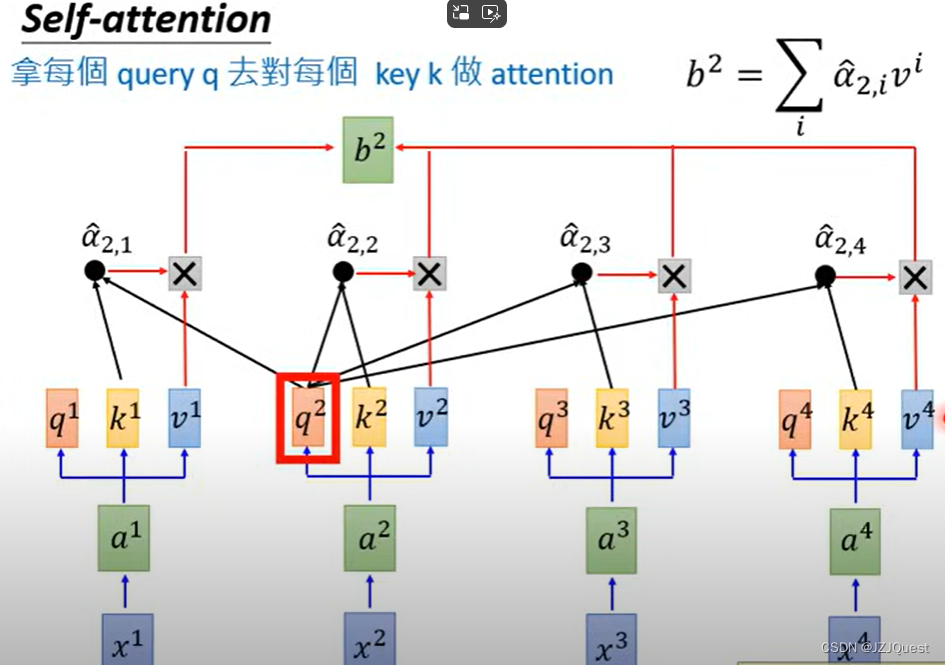
化为矩阵和向量的形式计算如下: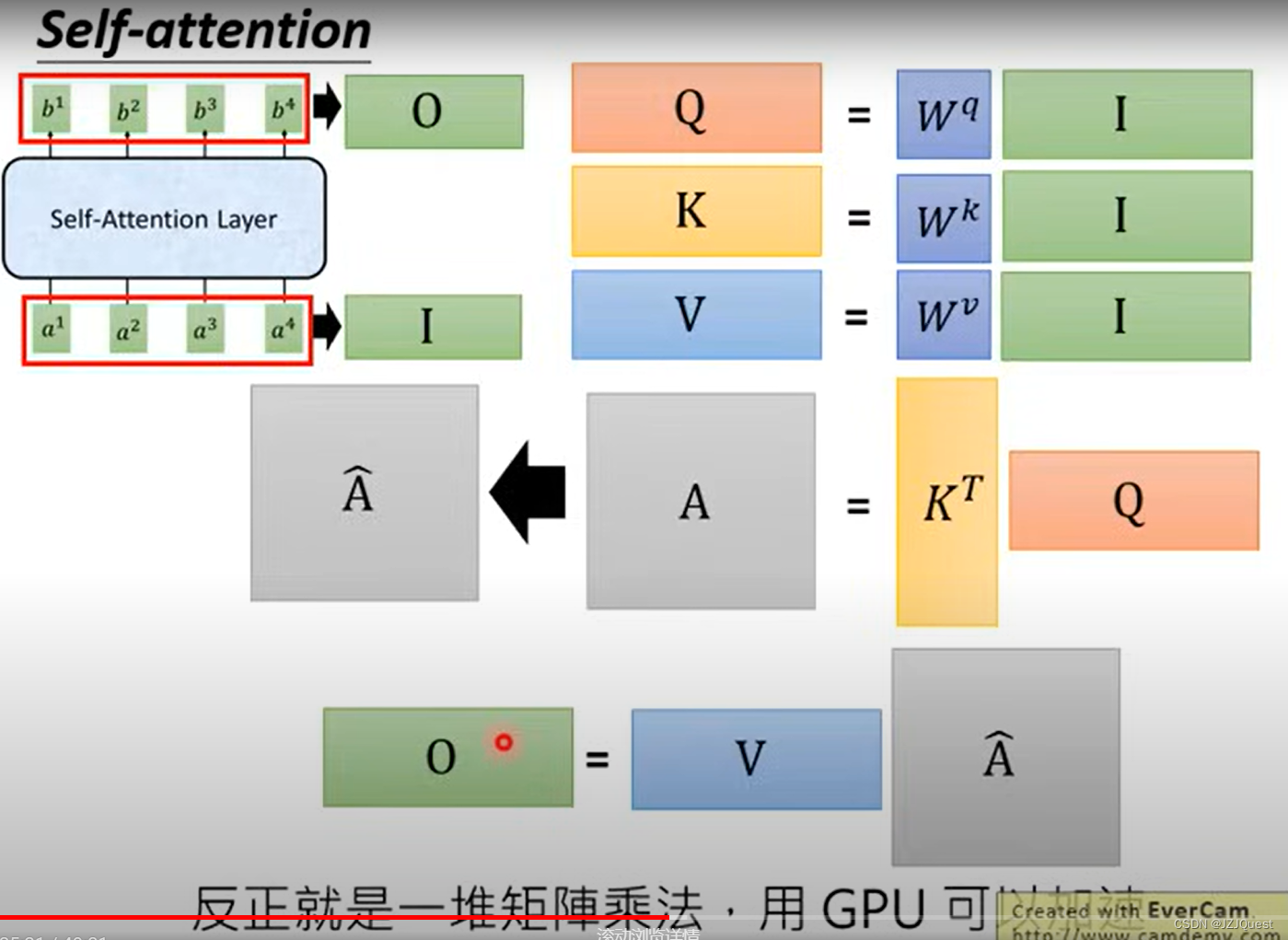
多头注意:同一组输入嵌入不同的参数中。
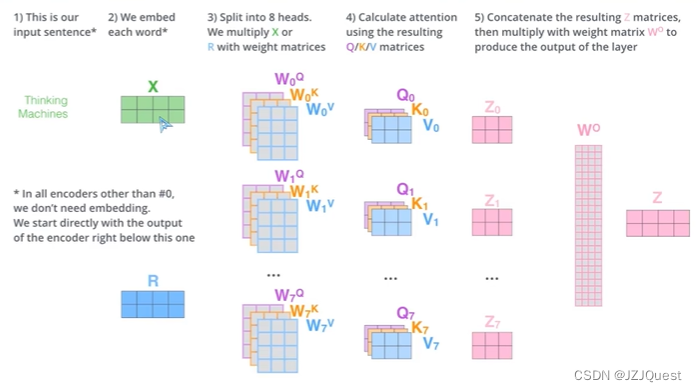
然后,在计算注意权重后,采用标度点积注意法[35]对相邻特征进行混合:
A
(
Q
i
,
l
,
K
i
,
l
,
V
i
,
l
,
R
i
,
l
)
=
φ
(
Q
i
,
l
,
K
i
,
l
,
R
i
,
l
)
V
i
,
l
,
(7)
A(\mathcal{Q}^{i,l},\mathcal{K}^{i,l},\mathcal{V}^{i,l},\mathcal{R}^{i,l})=\varphi\left(\mathcal{Q}^{i,l},\mathcal{K}^{i,l},\mathcal{R}^{i,l}\right)\mathcal{V}^{i,l},\tag{7}
A(Qi,l,Ki,l,Vi,l,Ri,l)=φ(Qi,l,Ki,l,Ri,l)Vi,l,(7)
计算注意力权重后,采用缩放点积注意力方法对相邻特征进行混合,也就是:
Attention
(
Q
,
K
,
V
)
=
softmax
(
Q
K
T
d
k
)
V
.
\text{Attention}(Q,K,V)=\text{softmax}(\frac{QK^T}{\sqrt{d_k}})V.
Attention(Q,K,V)=softmax(dkQKT)V.
缩放点积注意力是计算自注意力机制中每个代币的输出向量的常用方法。它通过对值进行加权求和来计算输出向量,其中权重是通过缩放查询和键之间的点积来获得的。 比例因子是关键维度 (dk) 的平方根,用于避免计算点积时出现梯度爆炸或消失问题。然后将softmax函数应用于缩放的点积,以获得注意力权重,该权重表示每个值向量在计算输出向量中的重要性。 可以使用矩阵乘法和张量运算有效地计算缩放的点积注意力,并且可以扩展以包含其他特征和约束,例如位置编码、掩蔽和层归一化。 在本例中,缩放的点积注意力用于混合相邻特征,这意味着每个标记的输出向量是通过对其相邻标记的值进行加权和来计算的。这允许模型捕获标记之间的依赖关系和交互,并计算更准确和更具表现力的表示。 综上所述,这句话描述了在计算注意力权重后,使用缩放点积注意力方法混合相邻特征的过程。缩放的点积注意力通过对值进行加权求和来计算输出向量,其中权重是通过在查询和键之间缩放点积来获得的。输出向量表示相邻令牌的混合特征,可用于进一步处理和预测。
φ
(
Q
i
,
l
,
K
i
,
l
,
R
i
,
l
)
=
s
o
f
t
m
a
x
(
Q
i
,
l
(
K
i
,
l
)
T
d
k
)
⊙
R
^
i
(
R
^
i
)
T
,
(8)
\varphi\left(\mathcal{Q}^{i,l},\mathcal{K}^{i,l},\mathcal{R}^{i,l}\right)=\mathrm{softmax}\left(\frac{\mathcal{Q}^{i,l}(\mathcal{K}^{i,l})^T}{\sqrt{d_k}}\right)\odot\hat{\mathcal{R}}^i(\hat{\mathcal{R}}^i)^T,\tag{8}
φ(Qi,l,Ki,l,Ri,l)=softmax(dkQi,l(Ki,l)T)⊙R^i(R^i)T,(8)
其中,
R
^
i
=
R
i
∥
R
i
∥
2
∈
R
N
i
×
3
\hat{\mathcal{R}}^{i}=\frac{\mathcal{R}^{i}}{\|\mathcal{R}^{i}\|_{2}}\in\mathbb{R}^{N_{i}\times3}
R^i=∥Ri∥2Ri∈RNi×3表示归一化的相对坐标,并
⊙
\odot
⊙表示元素乘法(element-wise multiplation)。
直观地说,在中心原子i的邻域中,当相对距离注意
(
Q
i
,
l
)
j
(
K
i
,
l
~
)
k
T
(\mathcal{Q}^{i,l})_{j}(\mathcal{K}^{i,\tilde{l}})_{k}^{T}
(Qi,l)j(Ki,l~)kT和相对坐标的归一化积
r
j
i
(
r
k
i
)
T
r
j
i
r
k
i
\frac{\mathbf{r}_{ji}(\mathbf{r}_{ki})^T}{r_{ji}r_{ki}}
rjirkirji(rki)T都具有高分时,相邻原子k可能与j高度相关。
然后,我们以残差的方式添加层归一化,最终在一个这样的注意层中获得自注意局部嵌入矩阵:
G
i
,
l
=
G
i
,
l
−
1
+
LayerNorm
(
A
(
Q
i
,
l
,
K
i
,
l
,
V
i
,
l
,
R
i
,
l
)
)
.
(9)
\mathcal{G}^{i,l}=\mathcal{G}^{i,l-1}+\text{LayerNorm}(A(\mathcal{Q}^{i,l},\mathcal{K}^{i,l},\mathcal{V}^{i,l},\mathcal{R}^{i,l})).\tag{9}
Gi,l=Gi,l−1+LayerNorm(A(Qi,l,Ki,l,Vi,l,Ri,l)).(9)
我们还尝试了其他与注意力相关的技巧,如预层标准化、多头注意力等。,带来的改善不大。实际上,如图1©所示,我们将此过程重复
l
(
l
≥
2
)
l(l≥2)
l(l≥2)次,以获得更完整的表示。
接下来,我们定义原子i的编码特征矩阵Di∈RM1 × M2:
D
i
=
(
G
^
i
)
T
R
~
i
(
R
~
i
)
T
G
˙
i
,
(10)
\mathcal{D}^i=\left(\hat{\mathcal{G}}^i\right)^T\tilde{\mathcal{R}}^i\left(\tilde{\mathcal{R}}^i\right)^T\dot{\mathcal{G}}^i,\tag{10}
Di=(G^i)TR~i(R~i)TG˙i,(10)
其中
G
˙
i
\dot{\mathcal G}^i
G˙i代表的
G
^
i
\hat{\mathcal G}^{i}
G^i子矩阵, 是取Gi的前M2(<M1)列。这里特征矩阵
D
i
D^i
Di,即描述符,保持了上面提到的所有不变性,其证明可以在参考文献1中找到。[7].然后,我们通过多层拟合网络传递,与中心原子的类型嵌入参数连接的重塑
D
i
D^i
Di:
e
i
=
e
(
D
i
,
T
i
)
.
(11)
e_i=e\left(\mathcal{D}^i,T_i\right).\tag{11}
ei=e(Di,Ti).(11)
然后给出系统的总能量作为
e
i
e_i
ei的总和,原子力
F
i
\mathcal F_i
Fi可以通过等式
(
1
)
(1)
(1)进一步计算。
3 模型(预)训练和微调
对于模型训练或预训练,我们对模型内的所有可训练参数w采用Adam随机梯度下降法[44],以最小化损失:
L
w
(
E
w
,
F
w
)
=
1
∣
B
∣
∑
t
∈
B
(
p
ϵ
∣
E
t
−
E
t
w
∣
2
+
p
f
∣
F
t
−
F
t
w
∣
2
)
.
(12)
\mathcal{L}_{\boldsymbol{w}}(E^{\boldsymbol{w}},\mathcal{F}^{\boldsymbol{w}})=\frac1{|\mathcal{B}|}\sum_{t\in\mathcal{B}}\left(p_\epsilon\left|E_t-E_t^{\boldsymbol{w}}\right|^2+p_f\left|\mathcal{F}_t-\mathcal{F}_t^{\boldsymbol{w}}\right|^2\right).\tag{12}
Lw(Ew,Fw)=∣B∣1t∈B∑(pϵ∣Et−Etw∣2+pf∣Ft−Ftw∣2).(12)
这里B表示小批量,|B|表示批量大小,t表示训练样本的索引。
E
w
E^w
Ew、
F
w
F^w
Fw表示模型输出,E、F是相应的DFT结果。我们还采用了一个调度器来调整训练过程中的前置因子
p
ϵ
p_\epsilon
pϵ和
p
f
p_f
pf,以在能量和力标签之间取得更好的平衡。如果可能的话,这里省略的维里误差可以加到训练损失中。
为了用新的数据集对预训练模型进行微调,我们首先用新数据集的新统计结果改变预训练模型最后一层的能量偏差,然后我们固定预训练模型中的部分参数并训练剩余的参数。在接下来的实验中,当只有类型嵌入参数固定时,我们获得了最佳性能。
3.1 回顾Adam
Adam原理
1.计算过去梯度的指数加权平均值,并将其存储在变量(使用偏差校正之前)和(使用偏差校正)中。
2.计算过去梯度的平方的指数加权平均值,并将其存储在变量(偏差校正之前)和(偏差校正中)中。
3.组合“1”和“2”的信息,在一个方向上更新参数
{ v d W [ l ] = β 1 v d W [ l ] + ( 1 − β 1 ) ∂ J ∂ W [ l ] v d W [ l ] c o r r e c t e d = v d W [ l ] 1 − ( β 1 ) t s d W [ l ] = β 2 s d W [ l ] + ( 1 − β 2 ) ( ∂ J ∂ W [ l ] ) 2 s d W [ l ] c o r r e c t e d = s d W [ l ] 1 − ( β 2 ) t W [ l ] = W [ l ] − α v d W [ l ] c o r r e c t e d s d W [ l ] c o r r e c t e d + ε \begin{cases} v_{dW^{[l]}} = \beta_1 v_{dW^{[l]}} + (1 - \beta_1) \frac{\partial \mathcal{J} }{ \partial W^{[l]} } \\ v^{corrected}_{dW^{[l]}} = \frac{v_{dW^{[l]}}}{1 - (\beta_1)^t} \\ s_{dW^{[l]}} = \beta_2 s_{dW^{[l]}} + (1 - \beta_2) (\frac{\partial \mathcal{J} }{\partial W^{[l]} })^2 \\ s^{corrected}_{dW^{[l]}} = \frac{s_{dW^{[l]}}}{1 - (\beta_2)^t} \\ W^{[l]} = W^{[l]} - \alpha \frac{v^{corrected}_{dW^{[l]}}}{\sqrt{s^{corrected}_{dW^{[l]}}} + \varepsilon} \end{cases} ⎩ ⎨ ⎧vdW[l]=β1vdW[l]+(1−β1)∂W[l]∂JvdW[l]corrected=1−(β1)tvdW[l]sdW[l]=β2sdW[l]+(1−β2)(∂W[l]∂J)2sdW[l]corrected=1−(β2)tsdW[l]W[l]=W[l]−αsdW[l]corrected+εvdW[l]corrected
3 实验
我们进行了大量实验来评估DPA-1的性能。首先,我们针对各种系统从头开始训练它,并与几种方案进行比较。然后,我们使用一个AlMgCu数据集来测试它在用单元素和二进制数据进行预训练后传输到三元系统的能力。最后,我们使用OC20数据集[36]中的OC2M子集预训练DPA-1,并将其应用于各种下游任务。为了说明类型嵌入和注意方案的有效性,我们在所有实验中与DeepPot-SE模型[7]进行了比较。在下文中,我们将首先介绍我们使用的数据集,然后介绍我们进行的实验。
我们进行了大量实验来评估DPA-1的性能。首先,我们针对各种系统从头开始训练它,
我们在简单的批量系统上从头开始训练DPA-1,并与嵌入式原子神经网络势(EANN)和DeepPot-SE进行比较。对于训练DPA-1和EANN,分别仅使用了10%和15∞20%的随机选择数据点,而DeepPotSE使用了90%。结果表明,即使训练样本较少,从头开始训练,DPA-1仍然基本上优于其他方法,特别是在力预测精度方面。
然后,我们使用一个AlMgCu数据集来测试它在用单元素和二进制数据进行预训练后传输到三元系统的能力。
对于AlMgCu、SSE和HEA系统,我们将它们分成具有不同成分的子集进行训练和验证。DPA-1和DeepPotSE的结果如表2所示。在训练损失几乎相同的情况下,DPA-1在验证准确性方面大大优于DeepPot-SE。
最后,我们使用OC20数据集[36]中的OC2M子集预训练DPA-1,并将其应用于各种下游任务。
github_代码练习部分
这段代码是一个 Python 脚本,它可以将一个 JSON 格式的配置文件转换为 YAML 格式的配置文件。下面是对代码的逐行解释:
- deepmd-kit/data/json/json2yaml.py
import argparse
import json
from pathlib import (
Path,
)
from warnings import (
warn,
)
这些是导入的 Python 库。argparse 库用于解析命令行参数,json 库用于读取 JSON 文件,Path 类是 pathlib 库中的一个类,用于操作文件路径,warn 函数用于发出警告信息。
def _main():
这是一个函数定义,名称为 _main。这个函数是整个脚本的入口点,它包含了整个脚本的主要逻辑。
parser = argparse.ArgumentParser(
description="convert json config file to yaml",
formatter_class=argparse.ArgumentDefaultsHelpFormatter,
)
这段代码创建了一个 argparse.ArgumentParser 对象,它用于解析命令行参数。description 参数指定了脚本的描述信息,formatter_class 参数指定了命令行帮助信息的格式化器。
jsons = list(Path.cwd().glob("*.json"))
jsons.sort(key=lambda x: x.stat().st_mtime, reverse=True)
jfile = jsons[0]
yfile = jfile.with_suffix(".yaml")
这段代码获取当前工作目录下所有的 JSON 文件,并将它们按照最后修改时间排序,取最新的一个作为默认输入文件。然后创建一个输出文件,它的名称是输入文件名称的副本,但扩展名改为 .yaml。
parser.add_argument(
"INPUT", default=jfile, type=Path, nargs="?", help="input json file"
)
parser.add_argument(
"OUTPUT", default=yfile, type=Path, nargs="?", help="output yaml file"
)
args = parser.parse_args()
这段代码定义了两个命令行参数,分别是输入文件和输出文件。default 参数指定了默认值,type 参数指定了参数的类型,nargs 参数指定了参数所对应的命令行参数的个数。然后调用 parse_args 函数解析命令行参数,并返回一个 argparse.Namespace 对象,它包含了解析后的命令行参数。
with args.INPUT.open("r") as infile, args.OUTPUT.open("w") as outfile:
这段代码打开输入文件和输出文件,分别用于读取 JSON 文件和写入 YAML 文件。
yaml.dump(json.load(infile), outfile, default_flow_style=False, sort_keys=False)
这段代码读取输入文件中的 JSON 内容,然后将其转换为 Python 对象,再将其写入输出文件中,但格式为 YAML。default_flow_style 参数指定了 YAML 输出的格式,sort_keys 参数指定了字典键的排序方式。
warn("The order of the keys won't be preserved!", SyntaxWarning)
warn("_comment keys will also be lostt in the conversion")
这两行代码发出了警告信息,提示用户注意输出文件中的字典键的顺序可能会发生变化,并且 _comment 键在转换过程中会被丢弃。
总结
在本文中,我们开发了DPA-1,这是一个基于注意力的深度电位模型,允许对原子数据集进行大规模预训练。我们从不同方面测试了DPA-1,显示了它在从头开始训练时在各种数据集上的准确性方面的优异性能,以及在用现有数据预训练时的样本效率。对类型嵌入参数的进一步研究表明了在OC2M上预训练的DPA-1的可解释性。
在未来,将训练数据集扩展到覆盖整个元素周期表将是令人感兴趣的,特别是在潜在空间中看到更收敛的“螺旋”;局部化学环境的包埋信息可能有助于表征不同的构象。多任务和无监督的培训计划值得探索;对于下游任务,就像在CV和NLP领域发生的那样,像模型压缩、提炼和转移等方案,都是迫切需要的。我们把这些可能性和更多的应用留给未来的作品。

















 390
390

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









