







2015年,计算机视觉在图像识别任务中首次超越人类视觉精度,这一里程碑事件被视为本轮人工智能技术爆发的标志性起点。随后,深度学习模型及其衍生算法呈现爆发式增长,在药物分子生成、材料性质预测、蛋白质结构预测等多个科学领域取得突破性进展。随着技术的持续演进,人工智能展现出前所未有的发展潜力。至2022年,以ChatGPT的横空出世为标志,人类正式迈入通用人工智能(AGI)探索的新纪元。人工智能已然成为当代科技发展中不可忽视的重要议题。本期专题将深入探讨人工智能技术在分子动力学模拟领域的进展及其影响。
AI 渗透到分子动力学模拟中的一些关键步骤
-
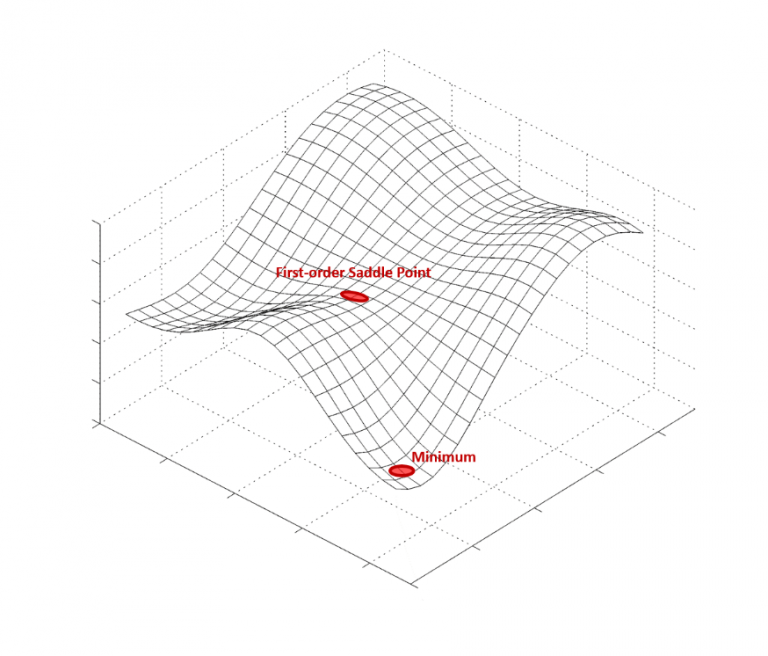
高效势能面(Potential Energy Surface, PES)构建
传统 MD 依赖经验力场(如 AMBER、CHARMM、OPLS 等),但 AI 尤其是深度学习(DL)和神经网络势(NNP)方法(如 DeepMD、SchNet、ANI)可以通过学习ab initio 数据(如 DFT、CCSD(T))构建高精度的势能面,从而在保持量子化学精度的同时显著提高计算速度。例如,DeePMD-kit 基于深度学习的势能面方法,已在蛋白质模拟、材料科学等领域展现了比传统力场更高的准确性和计算效率。
-
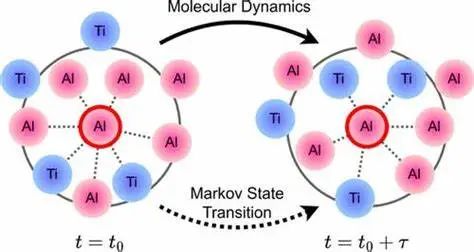
加速采样(Enhanced Sampling)
AI 可用于智能选择反应坐标并优化增强采样方法,如元动力学(Metadynamics)、自适应偏置力(ABF)和马尔可夫状态建模(MSM)。 强化学习(RL)方法可用于探索复杂自由能面,例如 AI-Guided Metadynamics 已被用于蛋白质折叠、配体结合等研究,提高了罕见事件的采样效率。
-
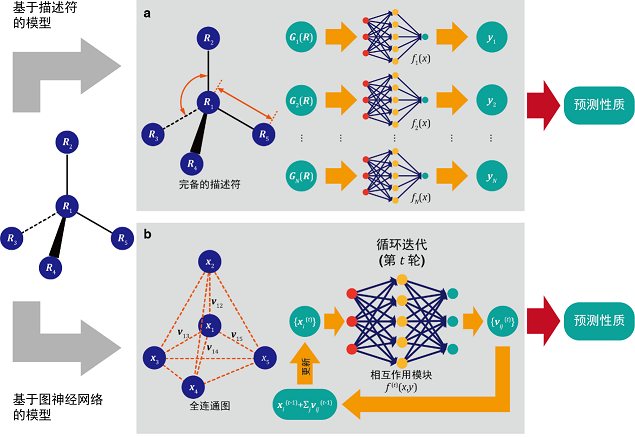
力场参数优化
传统力场参数化过程费时费力,AI 可自动优化参数,使其更符合高精度量子化学数据。例如,机器学习方法可以用于优化 Lennard-Jones 参数、静电势、二面角势能等,提高对特定分子系统的适用性。
-
分子构象预测与分析
机器学习方法(如 VAE、GANs、Transformer)可用于从 MD 轨迹中自动识别主要构象簇,进行自由能景观(FES)重建,并预测蛋白质折叠路径。 AI 结合图神经网络(GNN)可用于蛋白质-配体相互作用预测,甚至可以在 MD 轨迹数据的基础上预测化学反应路径。
-
数据驱动的MD模拟优化
AI 可用于从大量 MD 轨迹中提取关键特征,自动检测系统的动力学瓶颈,提高对蛋白质折叠、相变、化学反应等复杂过程的理解。例如,利用无监督学习(PCA、t-SNE)分析 MD 轨迹数据,以识别不同动力学状态。 基于 AI 的 MD 轨迹预测与生成
近年来,生成模型(如 Transformer、Diffusion Model)已被用于预测 MD 轨迹,并在小时间步长的情况下推测长时间演化行为,从而减少计算成本。
AI 在分子动力学模拟的未来发展方向
-
高精度端到端 AI 力场
未来 AI 可能会发展成为完整端到端的模拟框架,直接从量子化学数据学习势能面,并通过 AI-驱动的力场替代传统的经验力场,使模拟精度更接近 ab initio 计算,同时保留 MD 的时间尺度优势。 结合量子计算的 AI-MD 发展
量子计算结合 AI 可能彻底改变 MD 研究,尤其在电子结构计算、势能面拟合等方面,提高对大规模分子系统的模拟能力。

-
AI-Guided 多尺度模拟(Multiscale Modeling)
AI 可以智能地在全原子 MD、粗粒化 MD(CGMD)和连续介质模拟(如 Poisson-Boltzmann 方程)之间切换,提高计算效率,推动多尺度方法在生物大分子、材料等领域的应用。
-
自动化 AI-MD 平台
未来可能会出现全自动 AI-MD 平台,能够从头预测蛋白质折叠、配体结合、自组装过程,并提供类似 AlphaFold 的 MD 优化工作流。 AI 促进实验与模拟结合
结合 Cryo-EM、NMR、FRET 等实验数据,AI 可自动优化 MD 模拟,使其更符合实验观测结果,推动理论与实验的融合。
总结
AI已经在MD领域进行了广泛的应用,并将在高精度势能面构建、加速采样、力场优化、分子构象预测、自动化分析等方面持续推动MD方法的发展。有理由相信,AI与MD的深度结合将加速药物发现、生物物理等领域的研究,甚至可能完全改变传统MD模拟框架,使 AI-MD 成为下一代计算模拟的核心技术。




















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











