







 本文详细介绍了rdkit在药效团指纹(pharmacophore fingerprints)生成中的应用,包括其原理、具体实现和一个定制化案例。通过SMARTS定义原子类型和距离限制,构建了针对磷酸酯水解反应中(P,O)二元组特征的药效团策略。讨论了rdkit实现中的原子类型多样性、距离编码方式以及计数版本的可能性。
本文详细介绍了rdkit在药效团指纹(pharmacophore fingerprints)生成中的应用,包括其原理、具体实现和一个定制化案例。通过SMARTS定义原子类型和距离限制,构建了针对磷酸酯水解反应中(P,O)二元组特征的药效团策略。讨论了rdkit实现中的原子类型多样性、距离编码方式以及计数版本的可能性。
条件
基本熟悉 rdkit 的使用。
正文
概念
药效团指纹综合考虑了化学信息和结构信息,从原理上适合于描述分子间的相互作用。化学信息指的是利用SMARTS定义的一套原子类型的指定规则;结构信息指的是通过2D分子图上一对原子间的最短路径,为选取的原子对(二元组)、三元组、四元组等等引入几何信息。
下图(ref:10.1021/ci7003253)形象地说明了三元组的编码方案。值得注意的是,这篇文献的实现着重强调了同一个原子可能被分配于若干种原子类型,因此同样的原子组合可能贡献了若干指纹位。
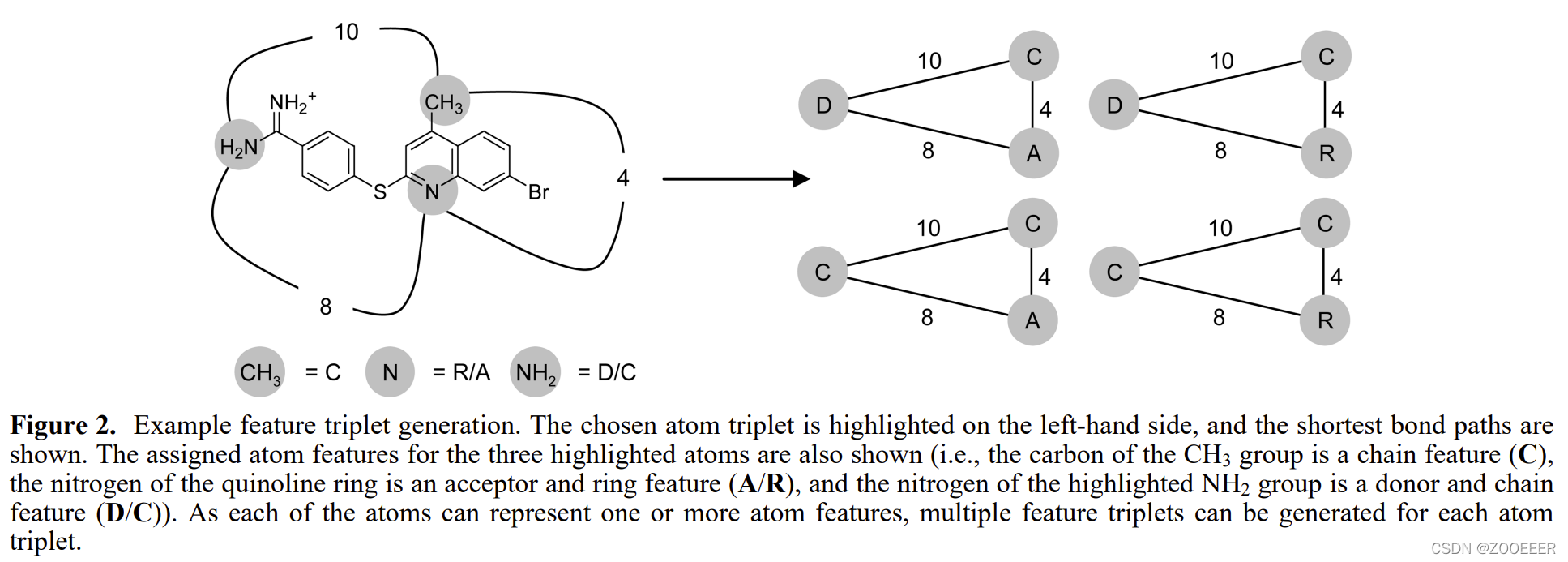
所有可行的组合:(参与原子数,原子类型,距离)(# point, patterns, distance bins)均作为一位(bit),形成具有固定长度的药效团指纹(pharmacophore fingerprints)。文档中做了清晰的图示,如下图。

实现
接口
按照文档实现一遍后,对接口应有了一个基本的了解。
这里会遇到的一个小问题是: .fdef 文件储存在哪里?
教程中表明可能在 (1) 下面的路径,或者 (2) 通过在 Python 库的文件夹中搜索 “fdef” 。
from rdkit import RDConfig
RDConfig.RDDataDir
BaseFeatures.fdef 中定义了27个AtomFeature,属于7个Family。MinimalFeatures.fdef 中定义了8个AtomFeature,属于5个Family。药效团特征是根据Family来做的。
进阶问题
若干值得考虑的问题:
- 该实现是否是 union 的,即是否支持一个原子的多种类型?
- 该实现是否可以实现计数版本,而非0-1版本?
- 对于四元组和五元组, rdkit 的实现中分别用5位和7位定义了距离,它指的是什么?是否合理?
案例:如何拓展药效团指纹的定义
在进入下面的案例前请阅读SMARTS的语法,fdef文件的语法。
面对特定的问题,可能想要定制特定的药效团策略。下面用一个案例来说明。
假设我们要处理磷酸酯水解反应,一般而言,磷酸酯水解发生在末端磷酸根基团上,例如ATP -> ADP -> AMP。我们想要定位这个末端的磷原子,并与主体结构的非磷酸根氧原子结合,做一个 (P,O) 二元组的距离分布特征。
Step 1 和 Step 2 完全是启发式的,只是为了探索一个合理的SMARTS定义策略。Step 3 写入了一个特征定义文件,然后基于若干真实化合物实现了上述策略。
Step 1:氧原子的环境
from rdkit import Chem
def evaluate(mols, patts):
for patt in patts:
hit_nums = []
for mol in mols:
matches = list(mol.GetSubstructMatches(patt))
hit_nums.append(len(matches))
print("hit_nums['{}']:".format(Chem.MolToSmarts(patt)), hit_nums)
patts = [
Chem.MolFromSmarts("O"),
Chem.MolFromSmarts("[O;H1]"),
Chem.MolFromSmarts("[O;D1]"),
Chem.MolFromSmarts("[O;D2]"),
Chem.MolFromSmarts("[O&!$(OP)]"),
Chem.MolFromSmarts("[O&!$(O~P)]"),
]
mols = [
Chem.MolFromSmiles("[P](O)(O)(=O)O"),
Chem.MolFromSmiles("CO"),
Chem.MolFromSmiles("C=O"),
Chem.MolFromSmiles("COC"),
Chem.MolFromSmiles("COP(O)(O)=O"),
]
evaluate(mols, patts)
输出如下:
hit_nums['O']: [4, 1, 1, 1, 4] # O原子
hit_nums['[O&H1]']: [3, 1, 0, 0, 2] # 羟基氧
hit_nums['[O&D1]']: [4, 1, 1, 0, 3] # 羟基氧和羰基氧
hit_nums['[O&D2]']: [0, 0, 0, 1, 1] # 醚基氧
hit_nums['[O&!$(OP)]']: [1, 1, 1, 1, 1] # 非P-OH氧
hit_nums['[O&!$(O~P)]']: [0, 1, 1, 1, 0] # 非P~O氧
Step 2:末端磷酸根中的磷原子
patts = [
Chem.MolFromSmarts("P"),
Chem.MolFromSmarts("[$(POC)]"),
Chem.MolFromSmarts("[$(POPOC)]"),
Chem.MolFromSmarts("[$(POPOPOC)]"),
Chem.MolFromSmarts("[$(P([O;H1])([O;H1])(=O)O)]"),
Chem.MolFromSmarts("[$(P([O;D1])([O;D1])(=O)[O;D2])]"),
]
mols = [
Chem.MolFromSmiles("CO"),
Chem.MolFromSmiles("OP(O)(=O)O"),
Chem.MolFromSmiles("COP(O)(=O)O"),
Chem.MolFromSmiles("COP(O)(=O)OP(O)(=O)O"),
Chem.MolFromSmiles("COP(O)(=O)OP(O)(=O)OP(O)(=O)O"),
]
evaluate(mols, patts)
输出为
hit_nums['P']: [0, 1, 1, 2, 3]
hit_nums['[$(POC)]']: [0, 0, 1, 1, 1]
hit_nums['[$(POPOC)]']: [0, 0, 0, 1, 1]
hit_nums['[$(POPOPOC)]']: [0, 0, 0, 0, 1]
hit_nums['[$(P([O&H1])([O&H1])(=O)O)]']: [0, 1, 1, 1, 1]
hit_nums['[$(P([O&D1])([O&D1])(=O)[O&D2])]']: [0, 0, 1, 1, 1]
Step 3:磷原子-氧原子距离特征
fdefName = "The_path_to_fdef_file"
content = """DefineFeature OxygenNotatPhosphate [O&!$(O~P)]
Family ONP
Weights 1
EndFeature
DefineFeature PhosphateAtEnd [$(P([O&D1])([O&D1])(=O)[O&D2])]
Family PAE
Weights 1
EndFeature
"""
with open(fdefName, 'w') as f:
f.write(content)
mols = [
Chem.MolFromSmiles("C(C(=O)O)OP(=O)(O)O"), # 0, Glycolic Acid O-P
Chem.MolFromSmiles("C([C@H]([C@H]([C@@H](C(=O)O)O)O)O)OP(=O)(O)O"), # 8, D-arabinonate-5-phosphate
Chem.MolFromSmiles("C([C@@H]1[C@@H]([C@@H]([C@H]([C@H](O1)O)O)O)O)OP(=O)(O)O"), # 87, D-galactose-6-phosphate
Chem.MolFromSmiles("C1=NC(=C2C(=N1)N(C=N2)[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(O)OP(=O)(O)OP(=O)(O)O)O)O)N"), # 107, ATP
Chem.MolFromSmiles("C([C@@H]1[C@H]([C@@H]([C@](O1)(COP(=O)(O)O)O)O)O)OP(=O)(O)O"), # 161, D-Fru-1,6-BP
]
featFactory = ChemicalFeatures.BuildFeatureFactory(fdefName)
sigFactory = SigFactory(featFactory,minPointCount=2,maxPointCount=2)
sigFactory.SetBins([(3,4),(4,5),(5,6),(6,7),(7,8),(8,9),(9,10),(10,11),(11,12),(12,13),(13,14)])
sigFactory.Init()
# sigFactory.GetSigSize()
bits = np.zeros((len(mols),sigFactory.GetSigSize()))
for i,mol in enumerate(mols):
from rdkit.Chem.Pharm2D import Generate
fp = Generate.Gen2DFingerprint(mol,sigFactory)
bits[i,fp.GetOnBits()] = 1
# 一共有11个bin,总共有33bit, bit:11-22是我们所需的P-O距离特征。
POs = pd.DataFrame(bits.T[11:11+11], index = [sigFactory.GetBins()[i] for i in range(11)])
通过下面的代码来目视检查结果:
i = 0 # 1,2,3,4
print(POs[i])
mols[i]
最后的结果:
一些对于结果的说明:
- 计算的是P与O之间的最短路径,可以正确处理环状结构;
- bin的写法是左闭右开。例如(3,4)指的是键长为3。

















 4860
4860

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









