






在生态学、统计学和生物学等领域,排序分析是一种用于探索和展示数据结构的多元统计技术。这种分析方法通过将多维数据集中的样本或变量映射到低维空间,以便更容易理解和可视化数据之间的关系。排序分析常用于研究物种组成、生态系统结构等生态学和生物学问题。
一、常见的排序分析方法:
-
主成分分析(Principal Component Analysis,PCA): 用于降维和识别主要的数据变异方向。适用于线性关系强烈的数据集,例如生态学中的物种丰富度或环境变量。
-
对应分析(Correspondence Analysis,CA):主要用于分析两个分类变量之间的关系。常用于分析生态学中的物种和环境因素之间的关系。
-
多维尺度分析(Non-metric Multidimensional Scaling,NMDS):用于非线性关系较强或者不适用于欧几里得距离的数据。适用于生态学中的生境相似性分析等问题。
-
典型对应分析(Canonical Correspondence Analysis,CCA): 类似于对应分析,但着重于解释数据中的结构,并通过最大化可解释的变异来找到约束的对应。适用于物种和环境变量之间的关系分析。用于分析两个表格(例如,物种数据和环境数据)之间的关系,结合了对应分析和多元回归的特点。
-
因子分析(Factor Analysis):用于识别隐藏在观测数据背后的潜在变量(因子),通常用于探索数据的内在结构。
二、经典排序分析方法的R实现
加载数据。
library(microbiome)
library(phyloseq)
library(ggplot2)
data(dietswap)
pseq <- dietswap
# Convert to compositional data
pseq.rel <- microbiome::transform(pseq, "compositional")
# Pick core taxa with with the given prevalence and detection limits
pseq.core <- core(pseq.rel, detection = .1/100, prevalence = 90/100)
# Use relative abundances for the core
pseq.core <- microbiome::transform(pseq.core, "compositional")用给定的方法和差异度量对样本进行投影。
# Ordinate the data
set.seed(4235421)
# proj <- get_ordination(pseq, "MDS", "bray")
ord <- ordinate(pseq, "MDS", "bray")Multidimensional scaling (MDS / PCoA)
plot_ordination(pseq, ord, color = "nationality") +
geom_point(size = 5)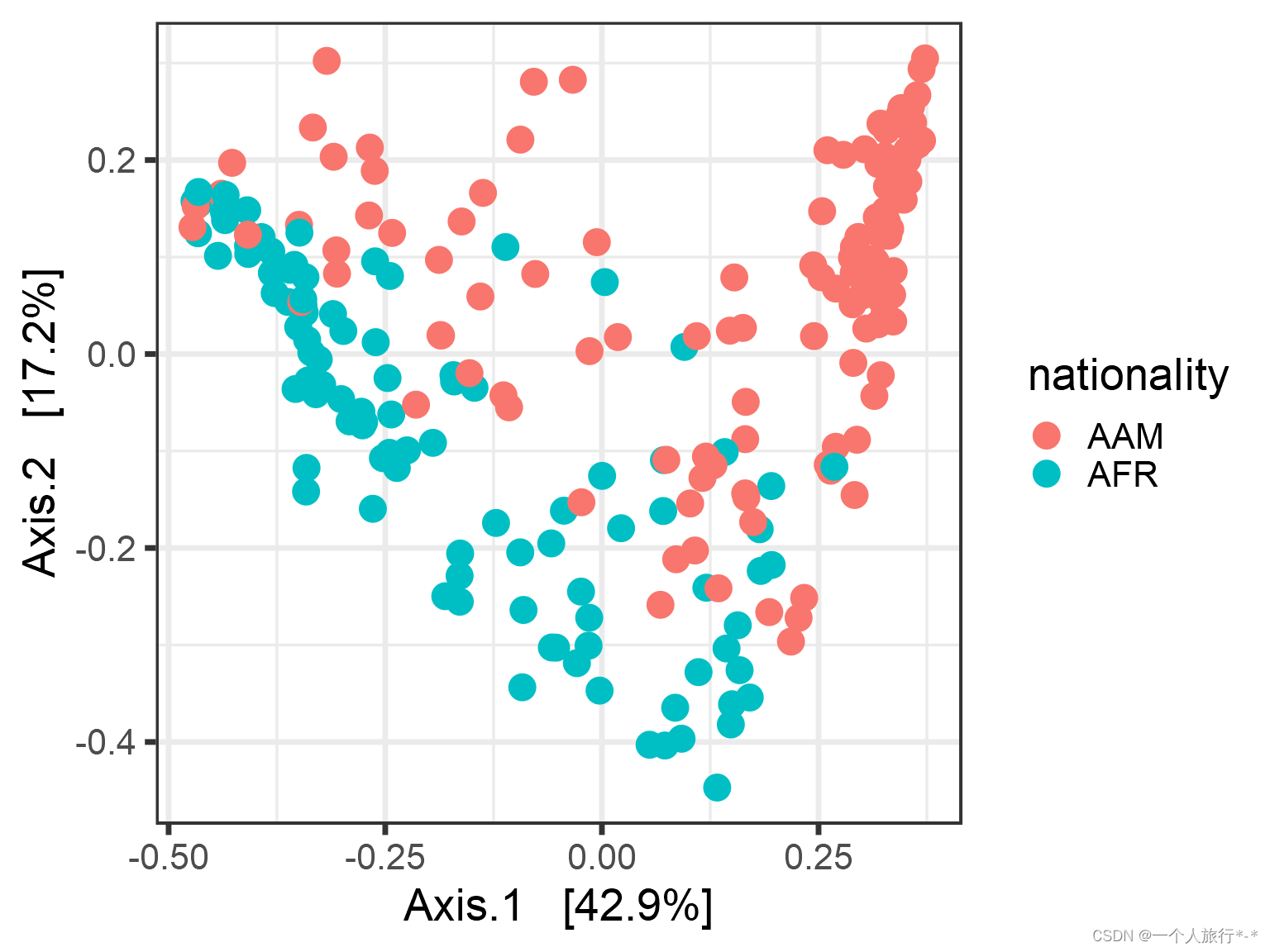
Canonical correspondence analysis (CCA)
# With samples
pseq.cca <- ordinate(pseq, "CCA")
p <- plot_ordination(pseq, pseq.cca,
type = "samples", color = "nationality")
p <- p + geom_point(size = 4)
print(p)
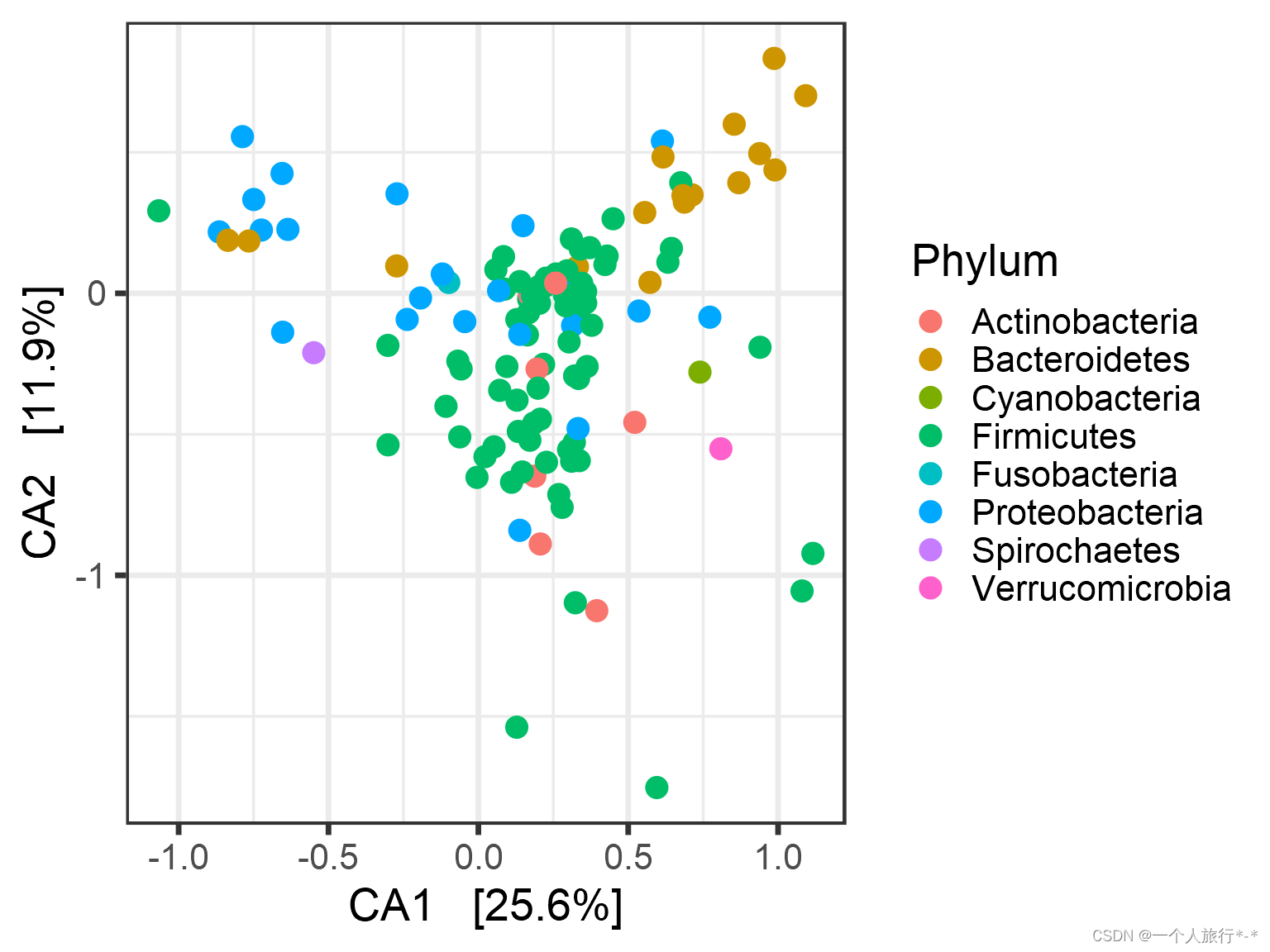
# With taxa:
p <- plot_ordination(pseq, pseq.cca,
type = "taxa", color = "Phylum")
p <- p + geom_point(size = 4)
print(p)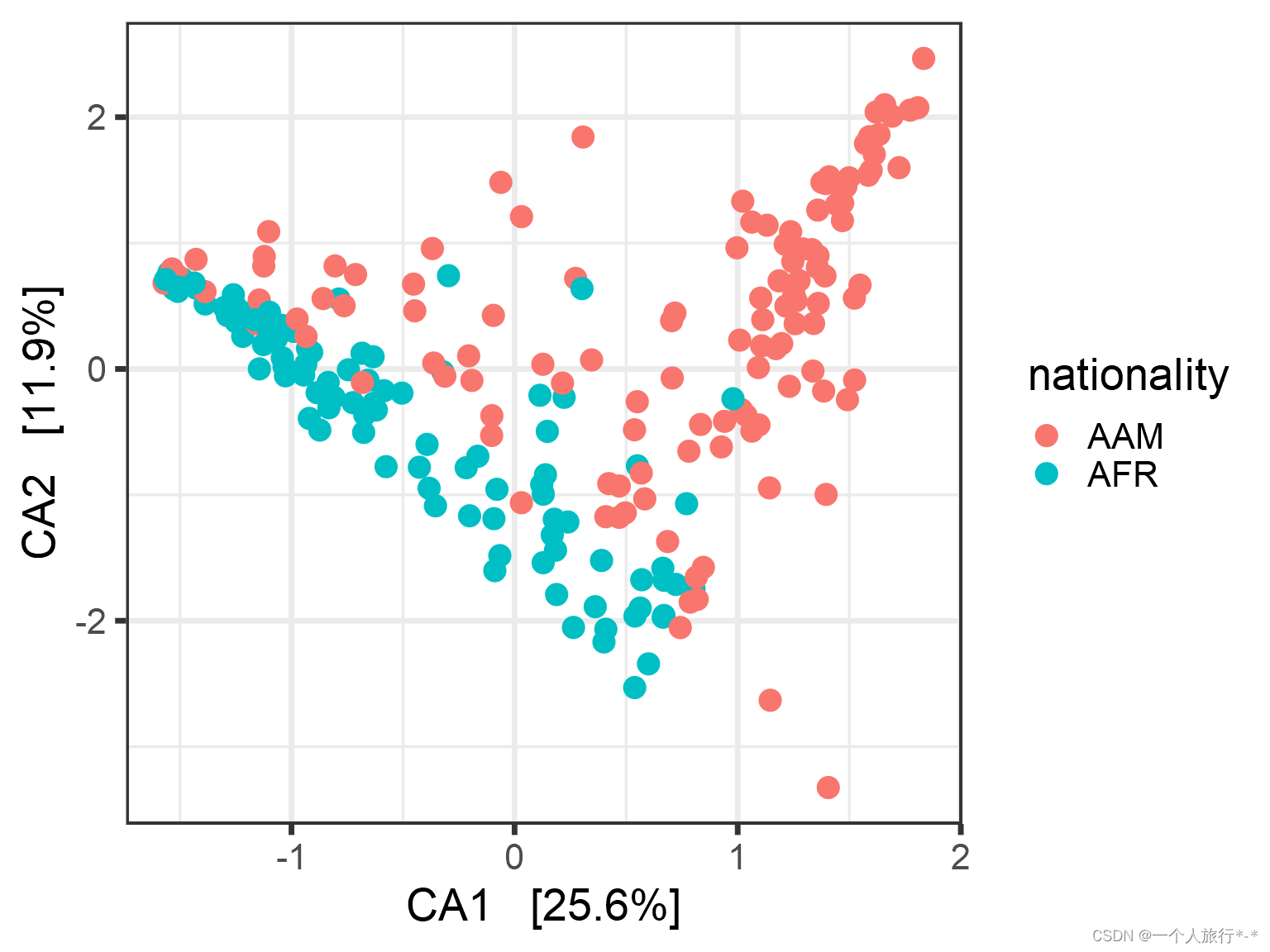
Split plot
plot_ordination(pseq, pseq.cca,
type = "split", shape = "nationality",
color = "Phylum", label = "nationality")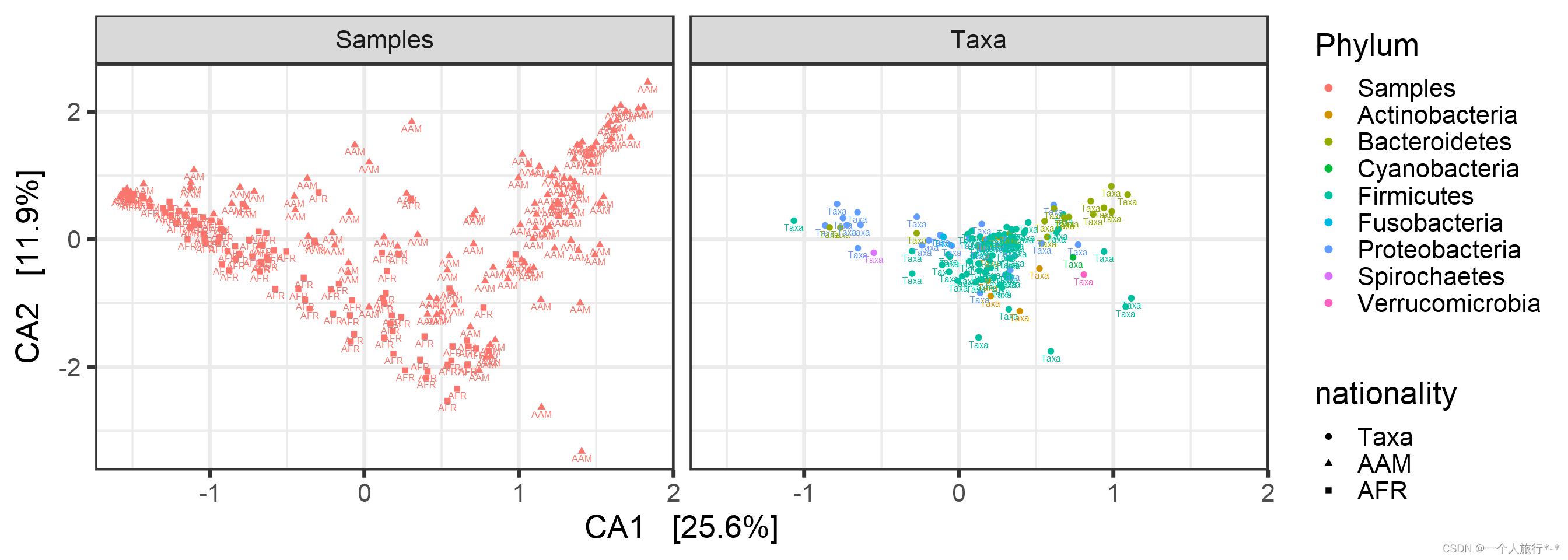
t-SNE
t-SNE是一种流行的新的排序方法。
library(vegan)
library(microbiome)
library(Rtsne) # Load package
set.seed(423542)
method <- "tsne"
trans <- "hellinger"
distance <- "euclidean"
# Distance matrix for samples
ps <- microbiome::transform(pseq, trans)
# Calculate sample similarities
dm <- vegdist(otu_table(ps), distance)
# Run TSNE
tsne_out <- Rtsne(dm, dims = 2)
proj <- tsne_out$Y
rownames(proj) <- rownames(otu_table(ps))
library(ggplot2)
p <- plot_landscape(proj, legend = T, size = 1)
print(p)
适用条件取决于数据的性质和研究的目标。在选择排序分析方法时,需要考虑数据的线性性、分布情况、相关性和可能存在的潜在结构。选择适当的排序分析方法有助于更好地理解数据集中的模式和关系。
References:















 2599
2599











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









