






一.生成cellchat文件,选择数据库
library(CellChat)
library(ggplot2)
library(ggalluvial)
library(svglite)
library(Seurat)
library(SeuratData)
#创建CellChat文件
cellchat <- createCellChat(object = sc, meta = sc@meta.data, group.by = "celltype")
#设置配体-受体相互作用数据库
CellChatDB <- CellChatDB.mouse #use CellChatDB.human if running on human data
showDatabaseCategory(CellChatDB)
cellchat@DB <- CellChatDB
#CellChatDB.use <- subsetDB(CellChatDB,
search = "Secreted Signaling",
key = "annotation") #也可选择指定数据库
cellchat <- subsetData(cellchat) 
二.处理表达数据
cellchat <- subsetData(cellchat)
#识别过表达基因
cellchat <- identifyOverExpressedGenes(cellchat)
#识别过表达配体受体对
cellchat <- identifyOverExpressedInteractions(cellchat)
cellchat <- projectData(cellchat,PPI.mouse)
#推断cell-cell communication network
cellchat <- computeCommunProb(cellchat,raw.use = F,population.size = TRUE)
cellchat <- filterCommunication(cellchat,min.cells = 10)
#计算每个信号通路相关的所有配体-受体相互作用的通信结果
cellchat <- computeCommunProbPathway(cellchat)
#计算整合的细胞类型之间通信结果
cellchat <- aggregateNet(cellchat)
三.可视化1:celltype之间通讯结果
groupSize <- as.numeric(table(cellchat@idents))
par(mfrow = c(1,2), xpd=TRUE)
netVisual_circle(cellchat@net$count, vertex.weight = groupSize,
weight.scale = T, label.edge= F,
title.name = "Number of interactions")
netVisual_circle(cellchat@net$weight, vertex.weight = groupSize,
weight.scale = T, label.edge= F,
title.name = "Interaction weights/strength")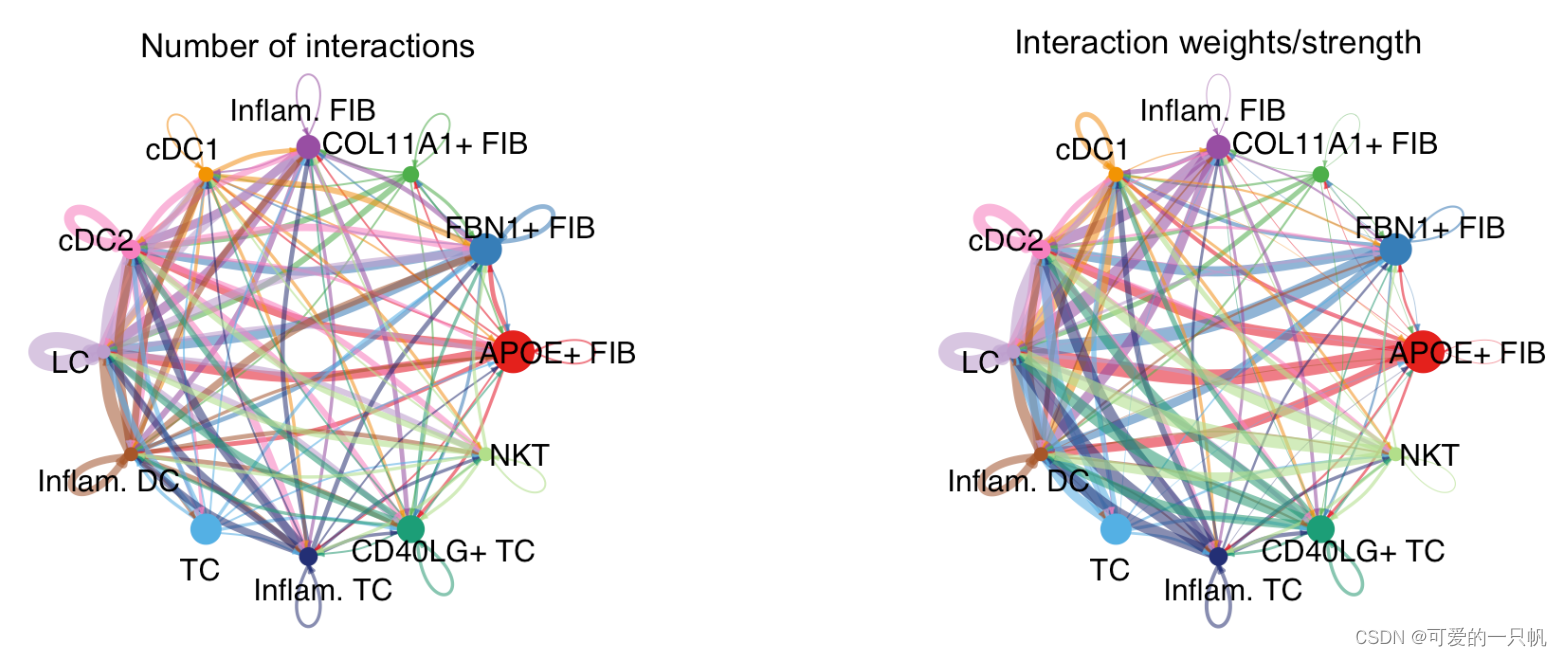
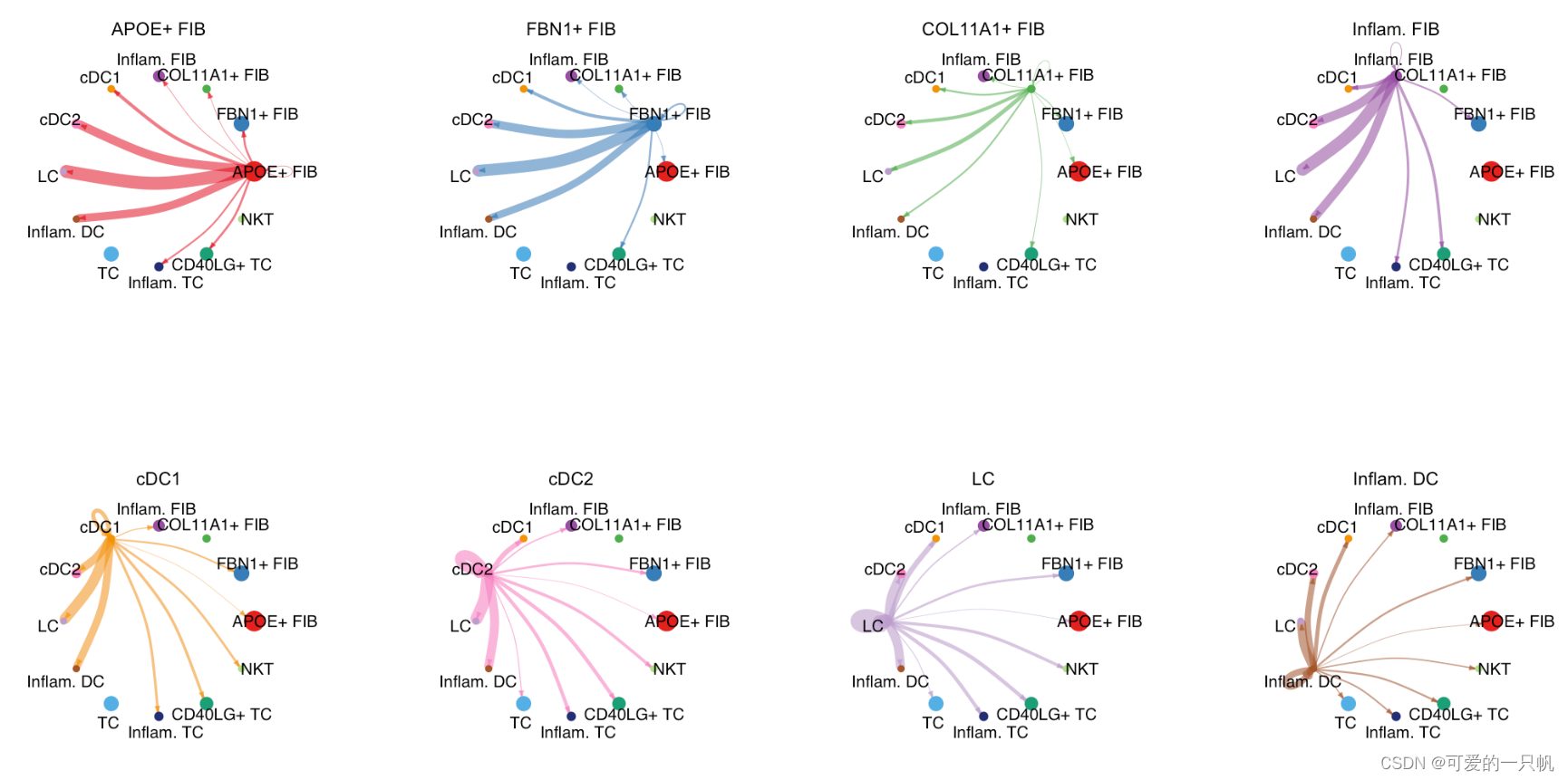
mat <- cellchat@net$weight
par(mfrow = c(3,4), xpd=TRUE)
for (i in 1:nrow(mat)) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat),
dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(mat2, vertex.weight = groupSize,
weight.scale = T, edge.weight.max = max(mat),
title.name = rownames(mat)[i])
}
可视化2:特定信号通路的互作情况
pathways.show <- c("CXCL")
netVisual_heatmap(cellchat, signaling = pathways.show, color.heatmap = "Reds")
可视化3:计算并可视化网络中心性分数
cellchat <- netAnalysis_computeCentrality(cellchat, slot.name = "netP")
netAnalysis_signalingRole_network(cellchat,
signaling = pathways.show,
width = 8, height = 2.5, font.size = 10)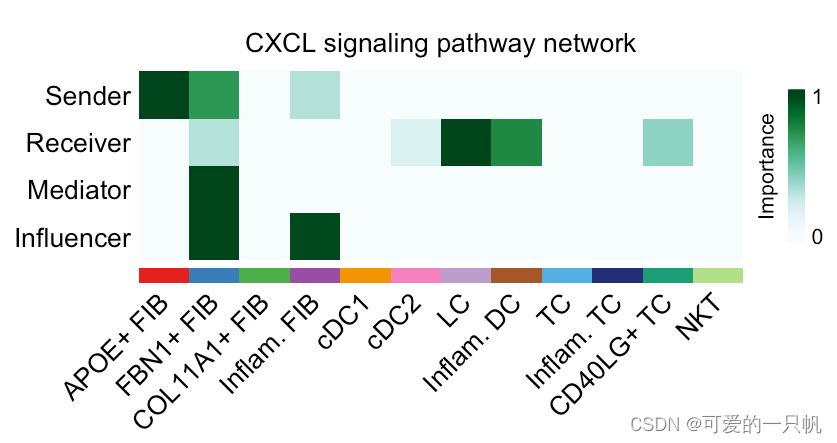
可视化4:根据结构相似性和功能相似性识别信号组
#功能相似性
cellchat <- computeNetSimilarity(cellchat, type = "functional")
cellchat <- netEmbedding(cellchat, type = "functional")
cellchat <- netClustering(cellchat, type = "functional")
netVisual_embedding(cellchat, type = "functional", label.size = 3.5)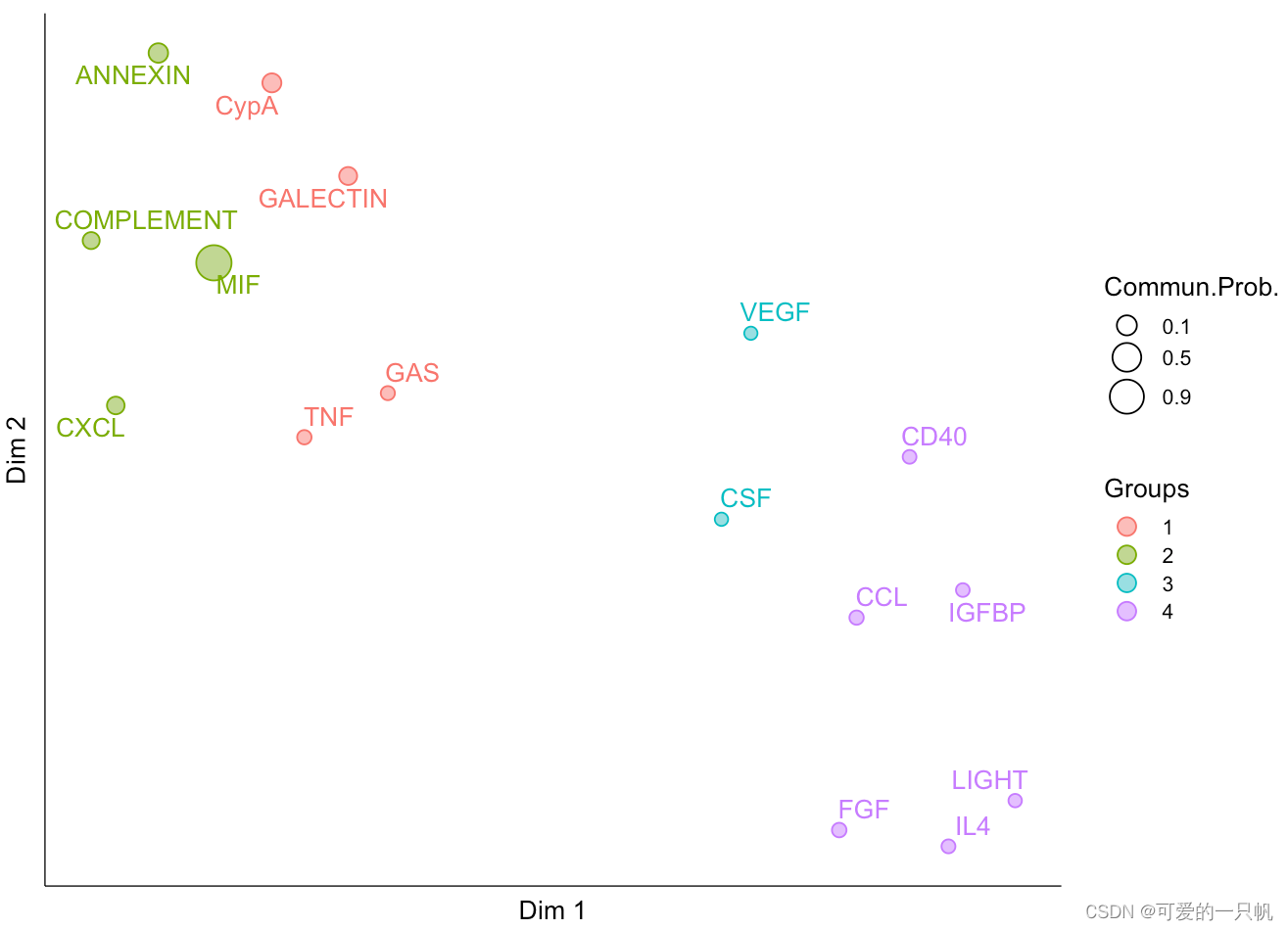
#结构相似性
cellchat <- computeNetSimilarity(cellchat, type = "structural")
cellchat <- netEmbedding(cellchat, type = "structural")
cellchat <- netClustering(cellchat, type = "structural")
netVisual_embedding(cellchat, type = "structural", label.size = 3.5)
netVisual_embeddingZoomIn(cellchat, type = "structural", nCol = 2)
参考:Inference and analysis of cell-cell communication using CellChat (htmlpreview.github.io)














 7391
7391











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









