







利用多组学整合鉴定人类疾病共享的和疾病特异性的宿主基因-微生物组关联
2022-05-23 本文系谷歌翻译
原文:Identification of shared and disease-specific host gene–microbiome associations across human diseases using multi-omic integration
网址:https://www.nature.com/articles/s41564-022-01121-z
编号:PaperTr-F001
摘要
虽然肠道微生物组和宿主基因调控独立导致胃肠道疾病,但尚不清楚两者如何相互作用以影响宿主病理生理学。在这里,我们开发了一个基于机器学习的框架来联合分析配对宿主转录组(n = 208)和肠道微生物组(n = 208) 来自结肠直肠癌、炎症性肠病和肠易激综合征患者的结肠粘膜样本的概况。我们确定了肠道微生物和宿主基因之间的关联,这些关联描绘了共享模式和疾病特异性模式。我们发现与胃肠道炎症、肠道屏障保护和能量代谢有关的一组常见宿主基因和途径与疾病特异性肠道微生物有关。此外,我们还发现与所有三种疾病有关的黏膜肠道微生物,如链球菌,与每种疾病的不同宿主途径相关,表明相似的微生物可以通过调节不同的宿主基因以疾病特异性的方式影响宿主的病理生理学。我们的框架可以应用于其他疾病,以识别可能影响疾病结果的宿主基因-微生物组关联。
Main
人类肠道微生物组在调节人类健康和疾病方面发挥着关键作用。人类肠道微生物组组成的变化与多种慢性疾病有关,包括结直肠癌 (CRC)、炎症性肠病 (IBD) 和肠易激综合征 (IBS)。例如,先前的研究报道了CRC中具核梭杆菌和细小单胞菌的丰度增加,减少了 CRC 和 IBD 中产肠毒素的脆弱拟杆菌的丰度和富集,以及肠杆菌科和IBD和IBS中的链球菌。除了肠道微生物组,宿主基因表达和通路的失调也与这些疾病有关。研究人员报告了 CRC 中 Notch 和 WNT 信号通路的破坏、诱导 IBD 中 NF-κB 和 TNF-α 信号通路的 Toll 样受体(例如,TLR4)的激活以及免疫反应失调IBS和肠道抗菌基因表达。虽然宿主转录和肠道微生物组已分别被确定为这些胃肠道 (GI) 疾病的促成因素,但尚不清楚这两者如何关联以影响宿主病理生理学。
对模式生物的研究表明,肠道微生物组对宿主基因表达的调节是微生物影响宿主生理的一种潜在机制。例如,在斑马鱼中,肠道微生物组负调控转录因子肝细胞核因子 4,导致宿主基因表达谱与人类 IBD 相关。在小鼠中,肠道微生物群可以改变宿主表观遗传程序以调节参与免疫和代谢过程的肠道基因表达。此外,最近的体外细胞培养实验表明,特定的肠道微生物可以改变相互作用的人类结肠上皮细胞中的基因表达。鉴于肠道微生物组和宿主基因调控之间存在串扰的证据,表征这两个因素之间的相互作用对于揭示它们在人类肠道疾病发病机制中的作用至关重要。
最近的一些研究调查了宿主转录组和肠道微生物组在特定人类肠道疾病(包括 IBD、CRC 和 IBS)中的关联。例如,检查 IBD 中微生物组-宿主基因关系的研究已经确定了粘膜微生物组与富含免疫炎症途径的宿主转录物的关联。在研究 IBD 中的纵向宿主-微生物组动力学时,Lloyd-Price 等人确定了趋化因子基因(包括CXCL6和DUOX2)的表达与肠道微生物(包括链球菌)丰度之间的关联和瘤胃球菌科。调查宿主基因-微生物组关联在 CRC 中的作用的研究发现,致病性粘膜细菌的丰度与与胃肠道炎症和肿瘤发生有关的宿主基因的表达之间存在相关性。在 IBS 中,与肠道屏障功能和肽聚糖结合有关的宿主基因,如KIFC3和PGLYRP1,与消化链球菌科和肠杆菌 8 的微生物丰度有关。虽然这些研究揭示了有关胃肠道疾病中宿主基因-微生物组串扰的重要见解,但它们在几个方面受到限制。例如,大多数研究都检查了有限的宿主基因子集与肠道微生物之间的关联。例如,仅关注差异表达的基因,与免疫功能相关的基因或选择代表细菌簇或共丰度组的微生物,因此仅表征潜在关联的子集。此外,宿主基因-微生物关联的识别基于使用 Spearman 或 Pearson 相关性测试每个宿主基因和微生物之间的成对相关性,因此忽略了这些数据集的固有多元属性。此外,大多数研究都集中在一次检查单一疾病的关联。因此,跨多种疾病状态的宿主-微生物组关联的常见和独特模式仍然缺乏特征。
在这里,我们全面描述了结肠直肠癌、炎症性肠病和肠易激综合征患者黏膜基因表达和微生物组组成之间的关联——三种胃肠道疾病,其中宿主基因调控和肠道微生物组都被认为是促成因素。 我们开发并应用了一个机器学习框架,该框架克服了多组学集成中的典型挑战,包括高维、稀疏和多重共线性,以识别每种疾病中肠道微生物与宿主基因和途径之间具有生物学意义的关联。我们利用我们的框架来描述三种疾病的疾病特异性和共享宿主基因-微生物组关联,这可能有助于深入了解这些胃肠道疾病病理生理学的分子机制。
结果
整合宿主基因表达和肠道微生物组丰度
为了研究跨疾病的宿主-微生物组关系,我们使用了从 CRC、IBD 和 IBS 患者获得的结肠粘膜活检产生的宿主基因表达 (RNA-seq) 数据和肠道微生物组丰度 (16S rRNA 测序) 数据(图1a)。对于我们研究中的每个人,我们获得了一对样本——一个微生物组样本和一个宿主基因表达样本。总的来说,在三个疾病队列中,我们的研究包括 208 对这样的微生物组和宿主基因表达样本(总共 416 个样本;补充表1)。除 CRC 的宿主基因表达 (RNA- seq ) 数据外,所有数据集先前已作为单独研究发表。有关疾病队列、样本、测序、质量控制和数据处理的详细信息,请参见方法。
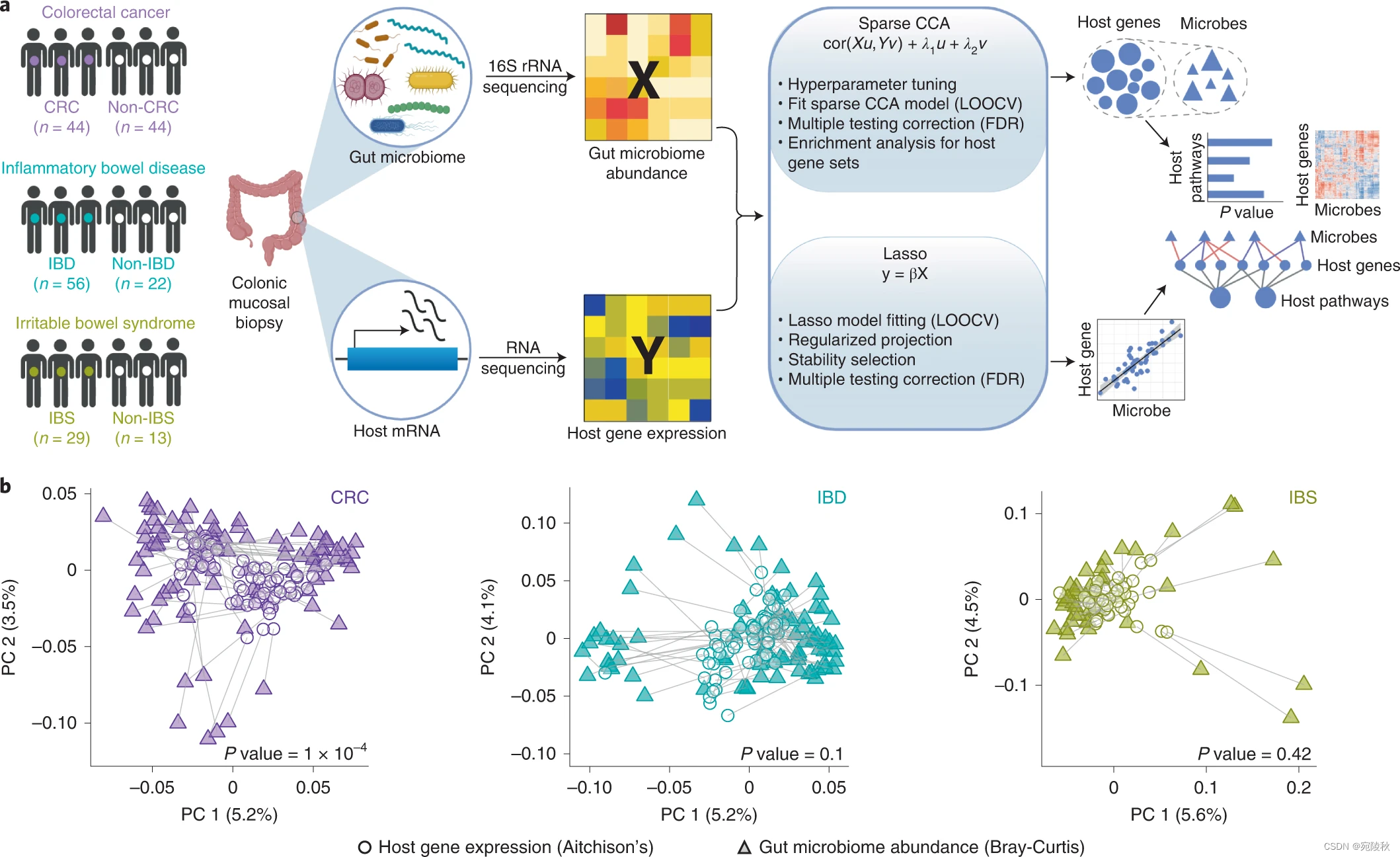
图 1:在 CRC、IBD 和 IBS 中整合宿主基因表达和肠道微生物组丰度。
a,研究设计概述:从每个疾病队列中的个体收集结肠活检样本,并为每个样本生成配对宿主转录组 (RNA-seq) 数据和肠道微生物组丰度 (16S rRNA) 数据。使用基于机器学习的框架整合配对的宿主转录组和肠道微生物组数据,以表征肠道微生物群与宿主基因之间的关联以及三种疾病的通路(从左到右)(有关整合框架和数学符号的详细信息,请参见方法)。
b, Procrustes 分析显示 CRC、IBD 和 IBS 中宿主基因表达变异与肠道微生物组组成之间的整体关联(从左到右)。我们将 Aitchison 距离用于宿主基因表达数据(圆圈),将 Bray-Curtis 距离用于肠道微生物组数据(三角形)。面板a是使用BioRender.com创建的。
以前的研究已经确定了人类肠道疾病中的宿主基因-微生物组关联,包括 CRC 、IBD和IBS 。因此,人们可能期望肠道基因表达模式和微生物组组成在这些疾病中具有广泛的相关性。为了测试宿主基因表达和肠道微生物组组成之间的这种整体关联,我们使用每个疾病队列的配对数据进行了 Procrustes 分析。我们的分析表明,在 CRC 中,宿主基因表达变异与肠道微生物组组成之间存在显着的对应关系(蒙特卡罗P值 = 0.0001)。然而,Procrustes 协议在 IBD(Monte Carlo P值 = 0.1)和 IBS(Monte CarloP值 = 0.42)(图1b和方法)。使用 Mantel 测试(方法)验证了这些结果。这种跨疾病宿主转录组和肠道微生物组之间缺乏显着的整体对应关系可能表明,很可能只有一部分肠道微生物与结肠上皮的一部分宿主基因相关,而不是两者之间的整体关联。因此,我们需要整合方法来表征这种宿主基因-微生物组的关联。
为此,我们使用 sparse canonical correlation analysis (sparse CCA, SCCA) 和 lasso penalized regression (套索回归)开发了一个基于机器学习的多组学集成框架。我们将这一方法应用于来自CRC、IBD和IBS的数据,以全面表征肠道微生物群与宿主基因之间的潜在生物学意义关联以及跨三种疾病的途径(图1,方法和扩展数据图1)。
CCA:典型相关性分析
Sparse CCA: Adaptive Estimation and Computational Barriers
Date: October 2017, DOI: 10.1214/16-AOS1519
lasso regression:套索回归
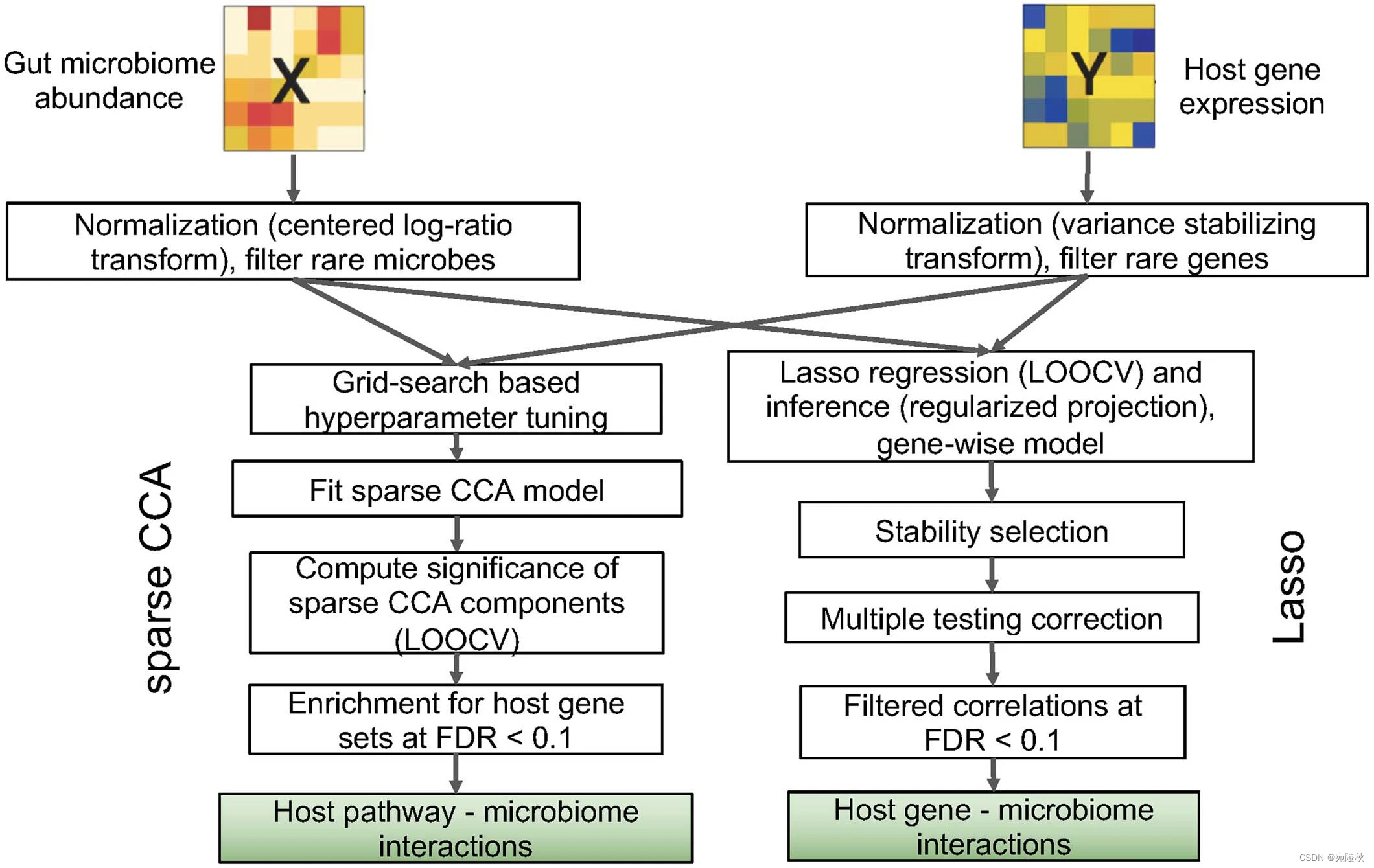
扩展数据 图 1:宿主基因-微生物组整合流程概述。
使用SCCA 和 lasso 方法整合肠道微生物组丰度和宿主基因表达数据的步骤。
共享宿主途径与疾病特异性肠道微生物有关
我们假设参与共同生物学功能的宿主基因和肠道微生物类群将以协调的方式发挥作用,因此将具有相关的表达和丰度模式。为了对此进行研究,我们使用SCCA 来表征三种疾病中宿主转录组和肠道微生物组之间的组级关联。我们为每个数据集拟合SCCA 模型,以识别显着相关的宿主基因和肠道微生物的子集,称为成分(方法和补充表2-4)。然后,我们对每个重要成分中的一组宿主基因进行了通路富集分析,以确定与疾病中肠道微生物相关的宿主通路。我们确定了“共享”途径,即基因表达与疾病队列中的肠道微生物相关的宿主途径,以及“疾病特异性”途径,即基因表达仅在三个疾病队列之一中与肠道微生物相关的宿主途径。图2a,Fisher 精确检验,Benjamini-Hochberg FDR < 0.1;补充表5)。为简单起见,我们专注于前五个最重要的共享和疾病特异性途径(图2a)。我们发现 CRC、IBD 和 IBS 共有三种途径,这些途径已知可调节胃肠道炎症、肠道屏障保护和修复。例如,已知作为线粒体能量代谢过程的氧化磷酸化在 IBD 和 CRC 中失调,并导致 CRC 中的肿瘤发生和耐药性。我们还发现疾病对之间存在重叠的宿主途径(参见图2a中的 CRC 和 IBD、CRC 和 IBS 以及 IBD 和 IBS ),包括免疫调节途径和细胞表面受体,如整合素途径、细胞和粘着斑以及蛋白酶体。
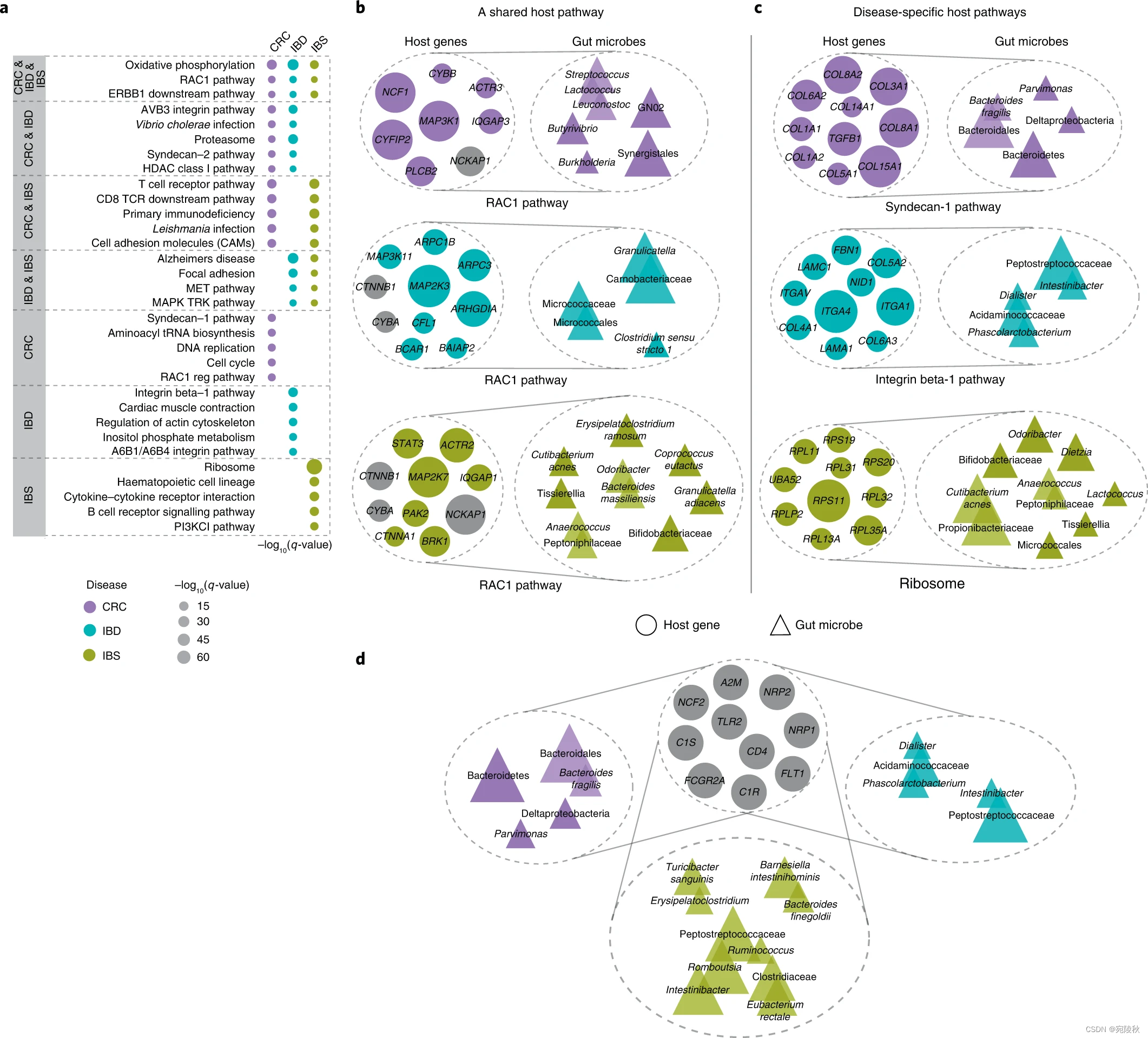
图 2:共享的免疫调节和代谢宿主途径与人类疾病中的疾病特异性肠道微生物相关。
a,宿主途径富含与疾病的肠道微生物组组成相关的SCCA基因组(FDR < 0.1)。点大小表示每个途径富集的重要性,点颜色表示该途径与微生物组组成显着相关的疾病队列。
b, CRC、IBD 和 IBS 中的微生物类群(从上到下)与 RAC1 途径中的宿主基因之间的关联,该途径中宿主基因表达与疾病队列中的肠道微生物相关(共享宿主途径)。圆形和三角形的大小分别代表基因和微生物的稀疏CCA系数的绝对值。属于常见分类顺序的微生物分类群显示为重叠三角形。至少两个疾病组的通路或成分之间共有的基因以灰色显示。
c,疾病特异性宿主途径中的宿主基因组(即基因表达与肠道微生物仅在一个疾病队列中相关的宿主途径)与 CRC、IBD 和 IBS 中的肠道细菌组之间的关联(顶部到底部)。
d,与疾病特异性微生物组相关的一组常见宿主基因(灰色圆圈)。这些宿主基因富含免疫调节和急性炎症反应途径。
此外,我们确定了与肠道微生物相关的 102 种疾病特异性宿主途径,包括我们研究队列中的 52 种 CRC 特异性、25 种 IBD 特异性和 25 种 IBS 特异性途径(补充表5和图2a)。虽然 IBD 特异性宿主途径包括 A6B1/A6B4 整合素途径和整合素 beta-1 途径,它们在 GI 炎症中调节白细胞募集,但 IBS 特异性途径包括免疫反应途径,包括 B 细胞受体信号传导途径和核糖体途径。
为了更好地了解作为共同关联基础的宿主基因-微生物关联,我们专注于 RAC1 途径,其中宿主基因表达与 CRC、IBD 和 IBS 中的微生物组组成相关。已知 RAC1 途径可调节免疫反应和肠黏膜修复,之前已涉及 IBD 和 CRC 图2b )。正如预期的那样,我们观察到三种疾病的这种共享途径的一些重叠宿主基因。然而,与它们相关的微生物类群是疾病特异性的。在 CRC 中,RAC1 途径与口腔细菌分类群有关,例如链球菌、Synergistales和GN02,其中链球菌已知物种与结直肠癌的发生有关。在 IBD 中,RAC1 宿主途径与先前与 IBD 相关的微生物类群有关,包括Granulicatella 38、39和狭义梭菌 1,这是一种与 IBD 相似的慢性肠病相关的微生物。在 IBS 中,该途径与细菌有关,例如已证明在结肠炎中普遍存在的Bacteroides massiliensis,以及已知在 IBS 中耗尽的双歧杆菌和臭味杆菌。
为了研究疾病特异性关联,我们考虑了宿主基因表达仅在三种疾病之一中与肠道微生物相关的独特宿主途径(图2c)。例如,我们发现仅在 CRC 中与肠道微生物分类群相关的 Syndecan-1 途径先前已被证明可调节癌细胞的致瘤活性。该途径中的宿主基因表达与微生物类群有关,例如小单胞菌属和脆弱拟杆菌,这些微生物类群已知会促进肠道癌变,并被认为是 CRC的生物标志物。 整合素-1 通路是 IBD 中疾病特异性宿主通路,被发现与消化链球菌科、肠杆菌和拟杆菌属相关,这些微生物类群已被先前的研究牵连到IBD中。为了评估不同疾病宿主基因成分的相似性,我们确定了一组在三种疾病成分之间共有的宿主基因,我们发现这些基因富含肠道上皮细胞中的免疫反应途径,包括血管内皮生长因子 (VEGF )、互补和凝血级联反应以及细胞因子-细胞因子受体结合(图2d ),Fisher 精确检验,Benjamini-Hochberg FDR < 0.1)。虽然这组宿主基因与特定疾病的微生物群有关,但我们还发现 IBD 和 IBS 之间存在重叠的微生物,例如消化链球菌科和肠杆菌 - 在胃肠道炎症中发现的大量微生物。
肠道微生物与个体宿主基因和途径有关
以前的研究表明,特定的微生物类群可以调节个体宿主基因的表达。因此,我们探索了每种疾病中个体宿主基因与肠道微生物之间的关联。为此,我们使用 lasso 回归模型来识别特定的肠道微生物类群,其丰度与宿主基因的表达相关。我们以基因方式拟合这些模型,使用每个宿主基因的表达作为响应变量,将肠道微生物类群的丰度作为预测因子。然后我们应用稳定性选择来识别稳健的关联(方法)。使用这种方法,我们分别在 CRC、IBD 和 IBS 中发现了 755、1,295 和 441 个显着和稳定性选择的宿主基因-分类群关联(图 2)。 FDR < 0.1)。这些代表 CRC 中 745 个宿主基因和 120 种肠道微生物之间的关联(补充表6),IBD 中 1,246 个宿主基因和 56 种肠道微生物之间的关联(补充表7),以及 IBS 中 436 个宿主基因和 102 种肠道微生物之间的关联(补充表8 和图3a )。特定宿主基因-微生物关联的例子可以在扩展数据图2 中找到。总体而言,我们观察到宿主基因-分类群关联中的疾病特异性模式。
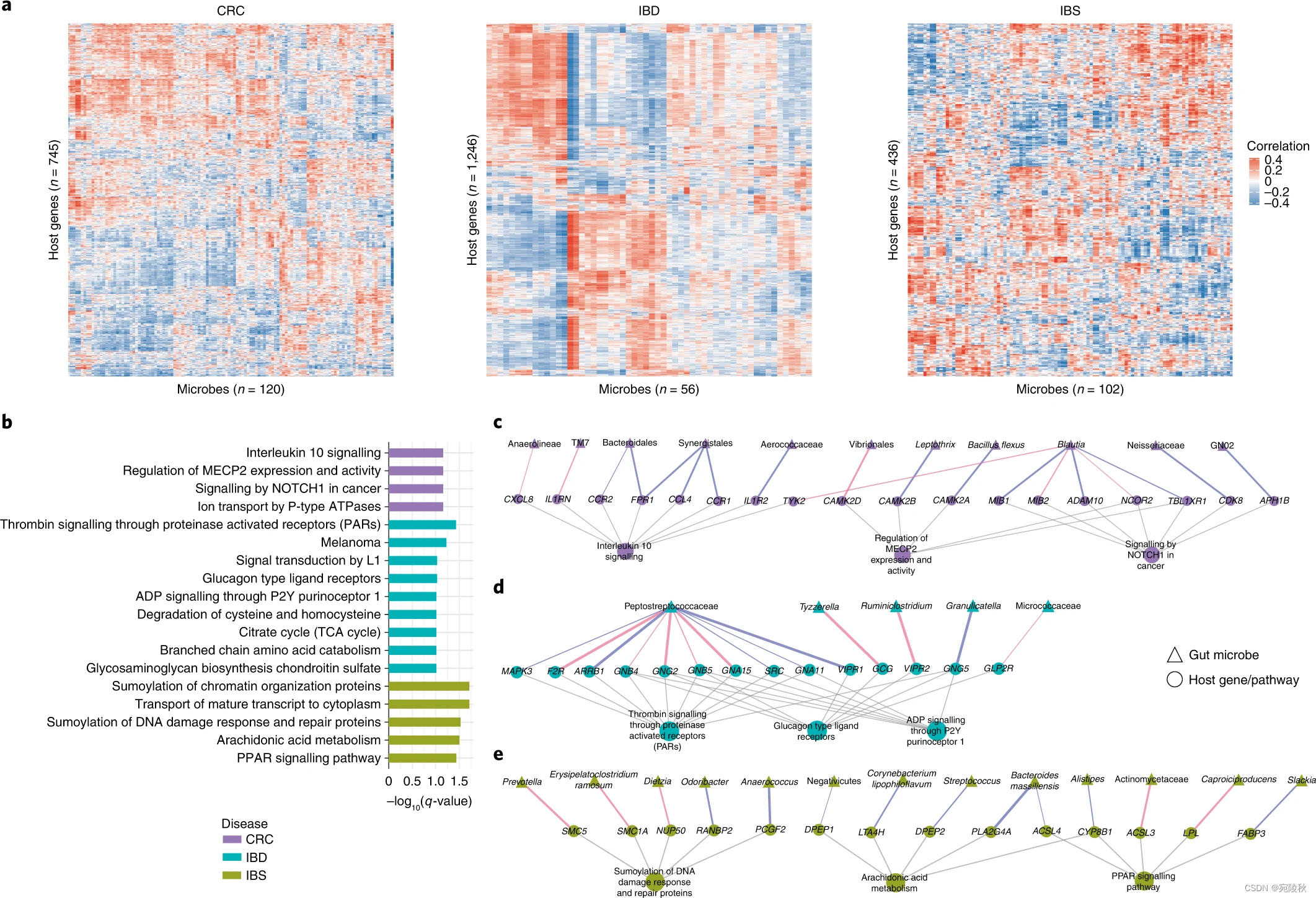
图 3:特定的肠道微生物与每种疾病中的个体宿主基因和途径相关。
a,热图显示了在 CRC、IBD 和 IBS (FDR < 0.1) 中通过套索模型识别的显着和稳定性选择的宿主基因 (行) 和肠道微生物分类群 (列) 之间的总体相关模式。
b,宿主通路在与 CRC(紫色)、IBD(绿色)和 IBS(黄色)中的特定肠道微生物相关的基因中富集(FDR < 0.1)。
c-e,网络显示特定肠道微生物与特定宿主基因相关,这些基因富集 CRC ( c )、IBD ( d ) 和 IBS ( e ) 中的疾病特异性宿主途径)。三角形节点代表肠道微生物,圆形节点代表宿主基因和途径。边缘颜色代表正(蓝色)或负(红色)关联,边缘宽度代表关联强度(Spearman rho)。灰色边缘代表宿主基因-通路关联。
为了表征与特定肠道微生物相关的宿主基因所代表的生物学功能,我们将富集分析应用于每种疾病中的一组肠道微生物群相关宿主基因(方法)。这与我们的组级方法(图2)互补,因为这些宿主途径在个体宿主基因-微生物对中富集。我们确定了每种疾病特有的 18 种宿主途径,包括 4 种 CRC 特异性途径、9 种 IBD 特异性途径和 5 种 IBS 特异性途径,这些途径与独特的肠道细菌相关(图3b,Fisher 精确检验,Benjamini-Hochberg FDR < 0.1;补充表9和方法)。已知在 CRC 特异性关联中富集的宿主途径可调节 CRC 中的肿瘤生长、进展和转移,例如白细胞介素 10 信号传导、癌症中的NOTCH1信号传导以及MECP2 表达和活性的调节。我们确定的在 IBD 特异性关联中富集的宿主途径已知负责维持胃粘膜完整性、炎症反应和宿主防御入侵病原体,例如通过蛋白酶激活受体 (PAR) 和胰高血糖素型配体受体的凝血酶信号传导。 对于 IBS 特异性关联,此处确定的丰富宿主途径已被证明可调节肠组织的稳态和 IBS 中的促炎机制,例如 DNA 损伤反应和修复蛋白的 sumoylation,以及花生四烯酸代谢。
为了表征宿主基因-微生物关联的潜在机制,我们进一步研究了与这些途径中的宿主基因相关的肠道微生物分类群(图3c-e)。在 CRC 中,我们发现 Anaerolineae 和 TM7(也存在于人类胃肠道中,已知可促进口腔和结肠直肠肿瘤发生的口腔微生物)与富含促肿瘤白细胞介素 10 的宿主基因呈负相关信号通路,例如CXCL8和IL1RN(图3c和扩展数据图2)。已知CXCL8在 CRC 和IL1RN中过表达主要参与免疫和炎症反应,其多态性与结直肠癌发生有关。白细胞介素 10 信号传导中的其他宿主基因,例如CCR2和FPR1,与拟杆菌目呈正相关(图3c和扩展数据图2)。CCR2和FPR1在结直肠肿瘤中过度表达,而拟杆菌在 CRC 中富集并与肿瘤发生相关。
我们观察到在IBD 患者中普遍存在的消化链球菌科与IBD中的多个宿主基因和通路相关(图3d)。例如,其丰度与宿主基因MAPK3和VIPR1的表达呈正相关,分别通过蛋白酶激活受体 (PAR) 和胰高血糖素型配体受体途径参与凝血酶信号传导。已知MAPK3在 IBD 的进展和发展中发挥作用,并且VIPR1在发炎的粘膜中过度表达。在 IBS 特定的关联中,我们发现了Prevotella的水平,已知在稀便的个体中过多,与 SMC5 的表达负相关,SMC5参与了 sumoylation 途径(图3e )。我们还发现PLA2G4A的表达,这是一种在花生四烯酸代谢中起重要作用并调节肠道上皮内稳态的宿主基因,与IBS中的Bacteroides massiliensis的丰度呈正相关,已知B. massiliensis在患者中普遍存在患有肠道恶性肿瘤(图3e)。总之,这些发现表明特定肠道微生物类群与特定宿主基因和途径之间的关联因疾病状态而异。
疾病特异性肠道微生物-宿主基因串扰
为了了解肠道微生物如何与疾病中的特定宿主基因相关联,我们探索了 CRC、IBD 和 IBS 中宿主基因-微生物关联之间的重叠(图4a,套索回归,Benjamini-Hochberg FDR < 0.1;补充表10)。我们确定了“共享”肠道微生物,即与至少两种疾病中的宿主基因相关的肠道微生物,并可视化了它们与跨疾病宿主基因的关联网络。我们发现了三种肠道微生物,消化链球菌科、链球菌和葡萄球菌,它们的丰度与我们研究队列中所有三种疾病的宿主基因表达相关(图4a,网络 1)。以前的研究表明,消化链球菌科和链球菌属 在CRC、IBD和IBS中发现水平升高。葡萄球菌属,虽然传统上被认为是鼻或皮肤相关细菌,但还定植于人类胃肠道,包括可能导致 CRC 和 IBD 患者急性肠道感染的机会性病原体,并与 IBS 和 CRC风险增加有关。我们发现消化链球菌科的丰度与宿主基因的表达呈正相关IBD中PYGB和NCK2与IBS中宿主基因HAS2的表达呈负相关。PYGB和NCK2在 IBD中均上调,其中已知PYGB可调节WNT /β-连环蛋白途径,并且NCK2参与整合素和表皮生长因子受体信号传导。相反,已知HAS2通过调节肠道稳态和炎症对结肠上皮细胞具有保护作用。 在 CRC 中,我们发现消化链球菌科的丰度与 GAB1 的表达呈负相关,GAB1是一种宿主基因,其过度表达会刺激结肠癌细胞中的肿瘤生长。在我们的研究队列中,链球菌还显示出与宿主基因表达相关的疾病特异性模式。在 CRC 中,其丰度与调节 WNT 信号传导和 NF-κB 通路的RIPK4的表达相关,并且在包括结肠癌在内的几种癌症类型中上调。同样,在 IBS 中,链球菌的丰度与DPEP2的表达相关,已知可调节巨噬细胞炎症反应(图4a,网络 1)。
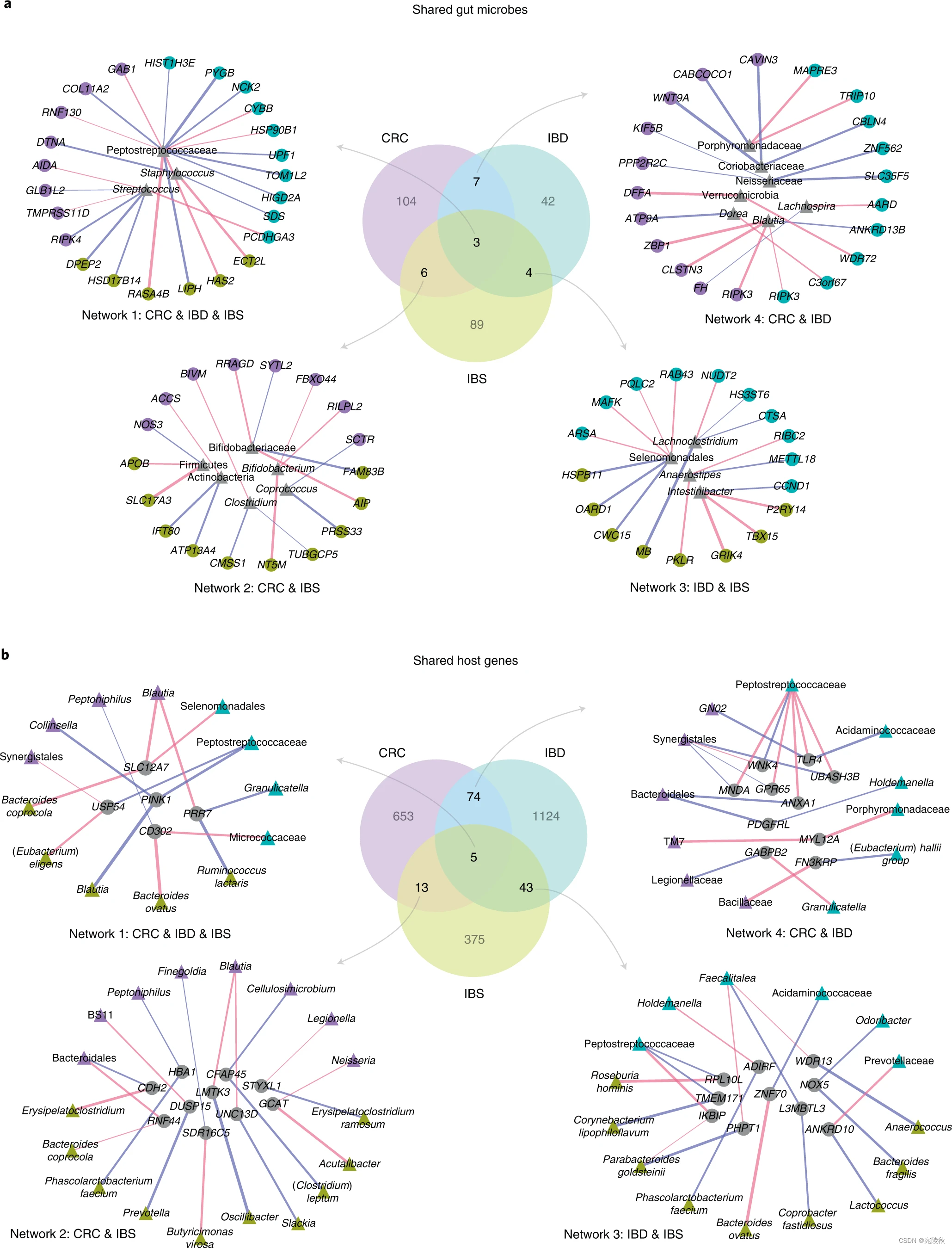
图 4:疾病特异性肠道微生物-宿主基因串扰。
a,“共享”肠道微生物的关联,即与至少两种疾病中的宿主基因相关的微生物。中:维恩图显示与 CRC、IBD 和 IBS 中的宿主基因相关的肠道微生物之间的重叠。逆时针,从左到右:显示肠道微生物宿主基因-微生物关联的网络在 CRC、IBD 和 IBS(网络 1)、CRC 和 IBS(网络 2)、IBD 和 IBS(网络 3)和 CRC 中共享和 IBD(网络 4)。
b,“共享”宿主基因的关联,即与至少两种疾病中的微生物相关的基因。中:维恩图显示与 CRC、IBD 和 IBS 中的肠道微生物相关的宿主基因之间的重叠。逆时针,从左到右:显示宿主基因-微生物关联的网络在 CRC、IBD 和 IBS(网络 1)、CRC 和 IBS(网络 2)、IBD 和 IBS(网络 3)和 CRC 中的关联中共享和 IBD(网络 4)。圆形节点代表宿主基因,三角形节点代表肠道微生物。彩色节点代表特定疾病(紫色,CRC;绿色,IBD;黄色,IBS),灰色节点代表肠道微生物(a)和宿主基因(b) 在疾病之间的关联之间共享。边缘颜色代表正(蓝色)或负(红色)关联,边缘宽度代表关联强度(Spearman rho)。所有关联均在 FDR < 0.1 时确定。
接下来,我们可视化了与两种疾病中的宿主基因相关的肠道微生物的宿主基因-微生物关联网络(图4a,网络 2-4,套索回归,Benjamini-Hochberg FDR < 0.1;补充表10)。我们发现了 20 种微生物,它们的丰度与至少两种疾病中宿主基因的表达有关。值得注意的是,发现产生丁酸盐的有益微生物Blautia的丰度与 CRC 和 IBD 中RIPK3的表达呈负相关(图4a,网络 4;扩展数据图3)。RIPK3促进 IBD 中的肠道炎症和结肠肿瘤发生。有趣的是,在 CRC 中,Blautia还与ZBP1(图4a,网络 4)相关,ZBP1 是一种宿主基因,可募集RIPK3以诱导 NF-κB 活化,并调节先天免疫反应以介导宿主对肿瘤和病原体的防御。
相反,为了探索相同的宿主基因如何与所有疾病中的不同肠道微生物相关联,我们确定了至少在两种疾病中与肠道微生物相关的宿主基因,并可视化了它们在疾病之间的关联网络(图4b,lasso 回归) ,FDR < 0.1;补充表11)。我们鉴定了 5 个这样的宿主基因,它们与 CRC 中的 4 种肠道微生物、IBS 中的 5 种肠道微生物和 IBD 中的 4 种肠道微生物相关(图4b,网络 1;补充表11)。值得注意的是,PINK1(一种调节线粒体稳态并激活 PI3 激酶/AKT 信号传导的宿主基因)的表达,有助于 IBD 中的肠道炎症和肿瘤发生 – 与CRC 中的 Collinsella、IBD 中的消化链球菌科和IBS 中的 Blautia的丰度有关。以前的研究发现,在 CRC中, Collinsella的丰度增加,而Blautia被发现与 IBS 症状呈正相关和负相关。
此外,我们确定了 135 个宿主基因,其表达与三种疾病中至少两种疾病中微生物类群的丰度相关,并可视化了最显着关联的网络(图4b,网络 2-4,套索回归,FDR < 0.1;补充表11)。我们发现在 CRC 和 IBD 中表达与肠道微生物相关的宿主基因富含参与免疫反应的途径,包括自然杀伤细胞介导的毒性、利什曼原虫感染和白细胞跨内皮迁移(图4b )。,网络 4,Fisher 精确检验,Benjamini-Hochberg FDR < 0.1)。这些宿主基因和与之相关的微生物分类群以前与CRC和IBD有关。例如,Annexin A1 或ANXA1(一种已知可调节肠黏膜损伤和修复的宿主基因,在 CRC 和 IBD 中发现失调)的表达与 CRC 中的拟杆菌属呈正相关,而与 IBD 中的消化链球菌科呈负相关(图4b,网络 4)。TLR4——一种已知通过识别细菌脂多糖来调节肠上皮炎症反应的宿主基因,以前与 IBD 和 CRC 有牵连 – 被发现与CRC 中的口腔微生物GN02相关,而在 IBD 中它与酸氨基球菌科相关 – 一种肠道微生物被发现在克罗恩病患者中大量增加(图4b,网络 4)。总体而言,我们的分析表明,在疾病关联之间共享的肠道微生物类群和宿主基因描绘了疾病特异性宿主-微生物串扰,因此表明宿主基因-微生物组关联的机制可能特定于疾病。
讨论
虽然肠道微生物群落和宿主基因表达分别与人类健康和疾病有关,但肠道微生物与宿主基因调控之间的关联在人类胃肠道疾病发病机制中的作用仍然很大程度上未知。使用基于机器学习的多组学整合框架,我们发现肠道微生物和宿主基因调控之间的共同和疾病特异性相互作用可能有助于胃肠道疾病的潜在病理生理学,包括 CRC、IBD 和 IBS。
以前的研究已经发现了 CRC、IBD 和 IBS 中常见的微生物特征。例如,所有三种疾病都表现出过多的消化链球菌科和链球菌属。此外,CRC 和 IBD 微生物组均表现为产生丁酸盐的肠道细菌(包括Blautia )的丧失,以及产肠毒素的脆弱拟杆菌的富集。与这些微生物组相似性相反,宿主基因调控在三种胃肠道疾病中表现出明显的变化;例如,独特的抗菌基因表达谱和嘌呤补救途径的破坏是 IBS 特有的,促炎性 IL-23/IL-17 信号传导的失调是 IBD 独有的,致癌途径(如 Notch 和 WNT 信号传导)的显着激活是CRC的标志。 在这里,我们发现相同的疾病相关肠道微生物可以与每种疾病中不同的宿主基因和途径相关联。因此,令人信服的假设是,尽管疾病可以以类似的微生物扰动为特征,但这些微生物可以通过与每种疾病中的不同宿主基因相关联来影响疾病特异性的病理生理过程。例如,我们发现在 CRC 中,链球菌与调节 WNT 信号传导和 NF-κB 通路的宿主基因的表达相关,而在 IBS链球菌中与调节巨噬细胞炎症反应的宿主基因相关,因此表明这种肠道微生物可能会扰乱 CRC 和 IBS 中不同的宿主途径。当然,由于我们的结果基于相关性分析,因此评估方向性具有挑战性。虽然这些疾病特异性关联可能在疾病发病机制中起作用,但疾病转化的结肠粘膜也可能使其更有利于相同的微生物类群。
我们还确定了一组常见的宿主基因和通路,这些基因和通路与所有三种疾病的肠道微生物组组成相关。这些包括调节胃肠道炎症、免疫反应和能量代谢的途径,并且以前与这些疾病有关。我们的分析表明,这些常见的宿主基因和通路与 CRC、IBD 和 IBS 中的疾病特异性肠道微生物相关。例如,我们发现调节 PI3 激酶/AKT 信号通路91的宿主基因PINK1的表达与 CRC 中的柯林氏菌、IBD 中的消化链球菌科和Blautia的丰度有关。在我们研究的 IBS 中。这表明在某些情况下,不同的肠道微生物可能会调节宿主基因和途径,这些基因和途径通常在不同的肠道病理中失调。同时,我们还发现了疾病特异性宿主基因-微生物关联。例如,在 CRC 中,Syndecan-1 通路是一种调节肿瘤生长和进展的宿主通路,它与已知会促进肠道癌变的微生物分类群如Parvimonas和Bacteroides fragilis相关。这些关联在 IBD 或 IBS 中没有发现,并且在我们的研究队列中是 CRC 独有的。宿主基因-微生物串扰的疾病特异性模式表明,肠道微生物,无论是通过与宿主细胞的直接相互作用还是通过间接相互作用(例如,通过产生特定代谢物),都可能在特定疾病背景下以不同方式调节宿主基因表达。
我们的研究有一些局限性。虽然我们报告了宿主基因-微生物组关联在胃肠道疾病的病理生理学中的潜在作用,但我们的研究确定了相关性,我们不能直接从这些结果中推断出因果关系。鉴于与研究人类因果机制相关的挑战,未来使用细胞培养或动物模型的研究将有助于阐明这些疾病中肠道微生物组与宿主基因调控之间关联的因果作用和方向性104。 此外,我们的分析仅关注微生物组的分类组成,因此我们无法表征涉及微生物基因和途径的关联。最后,有几个宿主和环境变量可能会影响微生物组和/或宿主基因表达,包括年龄差异、采样位置、饮食、宿主遗传学、治疗和用药史,这些在我们的疾病队列中是不可用的。因此,这些因素是可能影响我们结果的潜在混杂因素。
总体而言,我们的工作展示了整合肠道微生物组和宿主基因表达数据的能力,以深入了解它们在包括 CRC、IBD 和 IBS 在内的胃肠道疾病中的综合作用。总之,我们的结果表明,胃肠道疾病的特征在于微生物和宿主基因之间的复杂关联网络。尽管这些关联可能是特定疾病的,但我们发现相同微生物分类群与每种疾病中不同宿主基因相关的情况,反之亦然:相同宿主途径与每种疾病中不同微生物相关的情况。尽管微生物组研究中的许多努力都旨在确定导致疾病发病机制的特定微生物类群,但我们的研究结果表明整合宿主基因组学数据至关重要,因为它可以提供有关微生物影响健康的潜在机制的宝贵信息。我们的结果代表了朝着表征肠道微生物组与宿主基因调控之间的关联以及了解微生物组对疾病病因学的贡献迈出的重要一步。
方法
总体研究设计、样本和数据
总体而言,我们的研究包括 208 对微生物组 (16S rRNA) 和宿主基因表达 (RNA-seq) 样本,其中包括 CRC 队列中的 88 对样本(44 肿瘤和 44 患者匹配的正常),78对样本IBD 队列(56 名患者和 22 名对照)和42对 IBS 队列中的样本(29 名患者和 13 名对照)(补充表1和12-17)。除 CRC 的宿主基因表达数据外,所有数据集先前已作为单独研究发表。最初的研究获得了每个队列研究参与者的书面知情同意。在描述原始研究的出版物中可以找到有关数据收集过程中随机化和盲法的详细信息。没有使用统计方法来预先确定样本量,但我们的样本量与之前的出版物中报告的样本量相似。下面,我们详细描述 CRC 队列宿主 RNA-seq 数据的样本采集、测序和质量控制,并总结其他数据集的样本采集和数据处理和采集。
CRC 样本和数据
我们使用了来自 44 名患者的 88 对肠道微生物组和宿主基因表达样本,其中原发肿瘤和正常组织样本取自每个人。该队列中的个体包括 23 名女性和 21 名男性,平均年龄为 65 岁(中位数:67,范围:17-91)。Burns 等人对患者样本进行了表征和描述。简而言之,这些去识别的样本来自明尼苏达大学生物材料采购网络 (Bionet)。同时切除组织对,用无菌水冲洗,在液氮中快速冷冻,并由病理学家鉴定。该数据集的详细队列特征包含在补充表1中。
宿主 RNA-seq 测序、比对和质量控制
略
16S rRNA测序数据
略
IBD 样本和数据
略
IBS 样本和数据
略
预处理宿主基因表达数据
对于每个疾病队列的宿主基因表达数据,我们使用“biomaRt”R 包(版本 2.37.4)仅保留蛋白质编码基因的数据。我们过滤掉低表达基因以保留在每个疾病队列中至少一半样本中表达的基因。我们使用 R 包“DESeq2”(版本 1.14.1)对过滤后的基因表达读取计数数据120执行方差稳定转换. 我们过滤掉具有低方差的基因,使用每个疾病队列中样本间 25% 的方差分位数作为截止值。对每个疾病队列的 RNA-seq 数据分别执行这些步骤会产生一个独特的宿主基因表达矩阵,用于下游分析,包括 CRC 数据集中的 12,513 个基因、IBD 数据集中的 11,985 个基因和 IBS 数据集中的 12,429 个基因。
预处理微生物组数据
我们对每个疾病队列的微生物组数据分别执行了以下步骤。首先,如前所述,从OTU表中删除了被分类为源自古细菌、叶绿体、源自实验室试剂或试剂盒的已知污染物以及土壤或水相关环境污染物的序列。接下来,我们在物种(如果存在的话)、属、科、目、类和门的分类水平上总结了OTU表,并进行基于流行度和丰度的过滤,以保留至少10%样品中相对丰度为0.001的分类群。
为了允许在任何分类级别识别关联而不在每个分类级别重复分析,我们将不同级别的汇总分类单元矩阵组合成一个组合分类单元矩阵。这种方法可能会导致对分类组内的读数进行多次计数,从而导致在分类群丰度矩阵中添加相关特征。我们使用带有稳定性选择的套索的惩罚方法缓解了这个问题(参见方法:“套索回归分析”和“套索模型的稳定性选择”)。具体来说,这种方法不是从给定的分类进化枝中挑选多个相关的微生物分类群,而是仅从一组相关分类群中选择微生物分类群,其丰度与宿主基因的表达最密切相关。这种方法使我们能够识别在任何分类级别发现的信号,并通过将分析限制在单个分类级别来避免错过潜在的相关关联。同时,考虑到基因表达和微生物组数据等高维数据集中的大量特征,我们的方法避免了单独分析每个分类级别所需的计算密集型分析。
为了考虑微生物组数据集中的成分效应,我们测试了两种不同的方法来对每种疾病的分类数据执行中心对数比 (CLR) 转换:(1) 我们将汇总的分类矩阵(计数数据)连接成一个组合分类矩阵,然后对组合矩阵应用CLR变换,(2)我们对每个分类单元等级进行CLR变换,然后将它们连接成一个组合矩阵。我们验证了这两种转换方法是否相互关联。为此,我们比较了两种转换方法产生的类群丰度概况,发现这两种概况显着相关(P值 < 0.05),数据集中样本的平均 Pearson 相关性为 0.92,Spearman 相关性平均为 0.87(参见扩展数据图5例如,从 CRC 微生物组数据集中随机选择的几个样本的两次转换产生的分类群概况之间的相关性)。两种转化方法产生的分类群概况之间的这种一致性意味着转化方法不太可能影响下游结果。第一种方法生成的分类群概况在组成上是连贯的,并且在数据集中的分类群之间具有统一的转换,而第二种方法产生的分类群概况具有多峰分布(对应于每个分类等级的组成),这可能会使变量选择偏向于套索方法。因此,我们采用了第一种方法来转换我们的分类数据。
结果,我们获得了每个疾病队列的分类丰度矩阵,其中包括 CRC 数据集中的 235 个分类单元、IBD 数据集中的 121 个分类单元和 IBS 数据集中的 238 个分类单元。我们观察到,在 IBD 数据集中发现的独特分类群的数量是 CRC 和 IBS 数据集中发现的数量的 40%,因此这意味着 IBD 数据集的细菌多样性低于其他两个疾病数据集。这一观察结果与之前的研究一致,这些研究表明与没有这种疾病的个体相比,IBD 患者肠道黏膜微生物组的细菌多样性降低,包括生成和描述我们研究中使用的IBD数据集的HMP2研究。此外,以前的研究生成并表征了我们工作中使用的 CRC 和 IBS 数据集,报告称这些条件下的微生物多样性增加了。
跨疾病整合宿主基因表达和肠道微生物组数据
我们的研究包括三个不同的疾病队列,具有不同的样本收集、处理、制备和测序协议。先前的研究表明,无论使用何种数据处理管道,数据生成协议(包括样本收集、存储、DNA/mRNA 提取、PCR 扩增和测序)的差异都会导致潜在的批次效应。研究表明,即使使用相同的数据管理流程,数据生成流程中的偏差和批次效应仍然会影响分类组成和基因表达谱的分配。已经针对基因表达和微生物组数据集提出了校正批次效应的统计方法;然而,这些方法中的大多数都与检验病例和对照之间的差异有关,不适用于综合分析。
先前的研究还表明,与实验因素相比,不同的聚类方法,例如操作分类单元 (OTU)、零半径 OTU (zOTU) 和扩增子序列变体 (ASV),以及特定的管道设置可能对分类学分类没有重大影响例如测序引物的选择。为了在我们的数据集中检查这一点,我们使用 DADA2 管道重新分析了一些疾病数据集中的一些样本,它使用 ASV 聚类,并将结果与我们应用于处理我们研究中使用的数据的 OTU 聚类进行比较。我们发现估计的分类群概况在不同分类水平的两种方法之间是相关的。例如,在一些 CRC 样本中,我们发现使用 OTU 聚类和 ASV 聚类获得的分类群概况在不同分类水平上显着相关,包括在门(Spearman rho = 0.89,P值 = 0.0068)、类(Spearman rho = 0.68, P值 = 0.029) 和属 (Spearman rho = 0.6, P值 = 0.088) 水平。尽管有这些相当相关的分类概况,但由于下游分析的数据处理差异,很难评估总体偏差。此外,由于数据处理管道的差异导致的偏差与由于实验因素的差异导致的偏差很难解开和比较,因为后者在我们的研究中无法量化。这些综合差异可能会影响疾病队列中分类组成和基因表达谱的分配,进而影响下游整合分析。虽然很难完全消除这些偏差,但我们试图通过采用荟萃分析方法来最小化我们研究中疾病队列的整体批次效应,我们在其中进行了整合分析,并分别比较了每个队列中的疾病与对照样本,并在最后一个分析步骤中将跨队列的结果结合起来。虽然荟萃分析方法有缺点,例如降低统计能力,但在整合来自多项研究的基因组数据时,它们已被广泛用于最小化批次效应,并且最近在微生物组研究中被证明是有用的。
另一个潜在问题是疾病队列之间以及每个队列中病例组和对照组之间的样本量差异。在应用SCCA 和 lasso 回归时,这些样本量差异可能会导致统计功效的差异。这可能会影响每个疾病队列中确定的宿主基因-微生物关联和途径的数量,以及跨队列确定的重叠关联和途径的数量。我们试图通过应用差异富集分析来最小化这种影响,该分析由于样本量而对不同水平的统计能力更加稳健。此外,我们的分析仅关注微生物组的分类组成,因此我们无法表征涉及微生物基因和途径的关联。我们注意到,概括此处发现的一些结果可能具有挑战性,因为微生物组谱可能因个体和疾病而异。因此,研究微生物变化的功能库及其与宿主基因组数据的关联将是一个有希望的未来方向。
Procrustes 分析和 Mantel 测试
为了评估 CRC、IBD 和 IBS 中宿主基因调控和肠道微生物组组成之间的整体对应关系,我们使用“vegan”包(2.4-5 版)在 R 中进行了 Procrustes 分析。对于每个疾病队列,我们使用宿主基因表达数据的 Aitchison 距离和肠道微生物组数据的 Bray-Curtis 距离作为 Procrustes 分析的输入。使用具有 9,999 个排列的 ‘protest()’ 函数获得了轮换一致性的重要性。我们还应用 Mantel 测试来验证使用 R 中的 vegan 包(版本 2.4-5)在每个疾病队列中宿主基因表达的差异矩阵(Aitchison 距离)和肠道微生物组丰度(Bray-Curtis 距离)之间的整体相关性。我们还使用 Mantel 检验来验证每个疾病队列配对数据之间的总体对应关系,并发现与 Procrustes 分析的 CRC( P值 = 0.0026)、IBD(P值 = 0.2597)和 IBS(P值)相似的显着性模式= 0.9525)。使用 9,999 个排列来测试显着性。
集成框架概述
我们开发了一个机器学习框架,用于集成多组学高维数据集,例如宿主基因表达和肠道微生物组丰度。我们的整合方法有两个部分:(1)SCCA 用于识别与肠道微生物分类群相关联的宿主基因组,以表征通路水平的关联;(2)套索惩罚回归用于识别个体宿主之间的特定关联基因和肠道微生物类群(图1a和扩展数据图1)。我们将在下面详细描述这两种方法。我们将整合分析分别应用于每个疾病队列的配对宿主基因表达数据和肠道微生物组数据,以避免任何潜在的批次效应。对于每个疾病队列数据集,我们分别对患者数据(即 CRC、IBD 和 IBS)和相应的对照数据(分别为非 CRC、非 IBD 和非 IBS)进行整合分析,并仅考虑关联在患者中发现但在对照组中没有发现。在应用统计方法之前,我们对所有宿主基因表达和微生物组数据集中的数据进行了标准化和标准化,以满足统计模型的分布要求。
SCCA
SCCA模型的超参数调整和拟合
SCCA 分量的相关意义
SCCA 的富集分析
可视化来自SCCA 的疾病特异性和共享宿主途径和成分
套索模型的稳定性选择
在超级计算节点上并行执行 lasso 分析
套索输出的富集分析
比较病例与对照的关联和途径
识别和可视化在疾病关联中共享或不同的分类群和基因
以上省略
关于我们的方法的注释以及与以前研究的比较
据我们所知,虽然我们研究中使用的疾病队列先前已发表,但我们的研究似乎是第一个对宿主基因-微生物组串扰进行全面表征的研究。同伙。在之前描述CRC队列的研究中,Burns等人描述了肿瘤相关微生物组的特征,以及与相邻匹配正常结肠组织的微生物组相比,其组成如何变化。例如,他们报告了与正常样品相比,肿瘤相关微生物群中类杆菌目内多种分类群的丰度损失。在这里,我们发现拟杆菌目与宿主基因CCR2和FPR1相关,它们是肿瘤相关IL-10信号通路的一部分。先前描述 IBD 队列的研究比较了 IBD 与非 IBD 样本,发现属于梭菌纲的分类群之间的转录活性和丰度存在差异,以及疾病状态下免疫相关宿主途径的失调。我们对同一队列的综合分析揭示了免疫炎症通路与梭菌属内成员之间的关联,包括消化链球菌科和狭义梭菌 1。在描述 IBS 队列的原始研究中,Mars 等人。发现链球菌过多与健康个体相比,IBS 患者中的物种,并确定了粪便微生物(如消化链球菌科)与与肽聚糖结合有关的宿主基因之间的关联。我们的分析揭示了 IBS 中密切相互作用的组织粘附微生物组与宿主基因和途径之间的几个重要关联,包括链球菌与调节巨噬细胞炎症反应的宿主基因之间的关联,以及消化链球菌科与调节肠道稳态和炎症的宿主途径之间的关联。我们工作的一个重要贡献是基于机器学习的综合框架,用于表征人类疾病中的宿主基因-微生物关联。尽管最近很少有研究调查人类肠道疾病中宿主转录组和肠道微生物组之间的关联,但我们的分析使用了一种独特的分析技术,该技术具有几个优点。首先,与依赖于计算特征之间的成对相关性的分析相反(例如,Dayama 等人),我们的方法不需要将数据限制在预定的分类群或感兴趣的基因子集。此外,与通常用于查找配对数据集之间的整体对应关系的 Procrustes 分析相比,我们的方法不仅可以检测整体关联,还可以找到肠道微生物分类群与宿主基因(使用 lasso)和通路(使用稀疏的CCA),揭示了潜在的关联生物学机制。此外,我们的方法可以应用于其他类型的多组数据集,包括微生物代谢组和宏基因组数据8。最后,我们的分析结合了多种疾病的数据,确定了不同条件的共性以及特定疾病的模式。
我们的研究揭示了系统层面的关键见解;例如,我们发现与所有三种疾病(如链球菌)相关的肠道微生物与每种疾病中的不同宿主基因相关,这表明相同的微生物分类群可以通过潜在地调节不同宿主的表达来促成不同的健康结果结肠中的基因。我们还以疾病特异性关联的形式确定了许多特定假设;例如,我们发现: 拟杆菌属与大肠癌IL-10信号宿主通路中的宿主基因CCR2和FPR1相关;消化链球菌科与MAPK3和VIPR1相关它们是炎症性肠病中 G 蛋白偶联受体通路的一部分;和Bacteroides massiliensis与肠易激综合征中的宿主基因PLA2G4A相关,后者是前列腺素生物合成途径的成员。
数据可用性
CRC 队列的宿主 RNA-seq 的原始数据可在 BioProject ID PRJNA816986下的 NCBI 序列读取存档 (SRA) 上获得。之前公布的 CRC 队列 16S rRNA 测序的原始数据可在PRJNA284355 访问。以前发布的 IBD 队列的 16S rRNA 测序和宿主 RNA-seq 的原始数据可以分别在PRJNA398089和GSE111889访问。IBS 队列的 16S rRNA 测序和宿主 RNA-seq 的原始数据可分别访问PRJEB37924和GSE146853 . 每个疾病队列的宿主转录组学和微生物组数据的处理数据表已作为补充表包括在内(补充表12-17)。
代码可用性
本文中用于集成分析的代码可在
https://github.com/blekhmanlab/host_gene_microbiome_interactions
获得。我们还在
https://github.com/blekhmanlab/host_gene_microbiome_interactions/tree/main/Tutorial
中包含了我们的 integration pipeline 教程。
2022-05-23 小侯的大被窝
原文PDF下载:https://www.nature.com/articles/s41564-022-01121-z.pdf
补充表:https://static-content.springer.com/esm/art%3A10.1038%2Fs41564-022-01121-z/MediaObjects/41564_2022_1121_MOESM2_ESM.zip














 111
111











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









