







论文链接
https://www.sciencedirect.com/science/article/pii/S1470204523004680?via%3Dihub

一、Summary
1-1:Background
内容介绍了一项旨在提高肝细胞癌(HCC)(一种肝癌)特定治疗效果的研究。
这种治疗方法结合了两种药物:
- 阿特珠单抗(atezolizumab):一种免疫疗法,通过阻断PD-L1蛋白质发挥作用,这种蛋白质能帮助肿瘤躲避免疫系统;
- 贝伐珠单抗(bevacizumab):一种抗体,能抑制一种为肿瘤提供营养的血管生长所必需的蛋白质(抗血管生成疗法)。
研究发现,只有一部分 HCC 患者能从阿特珠单抗-贝伐单抗联合疗法中获益,这表明需要更好的患者选择标准来优化治疗效果。为了解决这个问题,研究人员重点开发了生物标志物,这是一种可用于预测患者对特定治疗反应的可测量指标。
阿特珠单抗-贝伐单抗反应特征(ABRS)是一种生物标志物,它与患者疾病在治疗期间和治疗开始后不再进展的时间长短有关,即所谓的**无进展生存期(PFS)。这项研究的主要目标是创建一个能够预测 ABRS 表达的人工智能模型,方法是分析组织样本或组织学切片的数字化图像**,这些图像经过染色并在显微镜下观察,以研究细胞结构的细节。
研究人员希望确定人工智能模型对 ABRS 表达的预测是否可以作为替代标记来估计患者的 PFS。如果成功,该模型可以帮助医生确定哪些患者最有可能从阿特珠单抗-贝伐单抗联合疗法中获益,从而实现个性化治疗,并有可能提高HCC患者的总生存率和生活质量。这是人工智能和机器学习如何融入临床决策以推进精准医疗的一个范例。
1-2:Methods
内容介绍了一项**回顾性多中心**研究的方法,该研究旨在开发一种名为 ABRS 预测(ABRS-P)的预测模型,用于估计肝细胞癌(HCC)患者的阿特珠单抗-贝伐单抗反应特征(ABRS)。ABRS 是与阿特珠单抗和贝伐珠单抗治疗患者无进展生存期 (PFS) 相关的生物标志物。
下面是研究方法的详细介绍:
模型开发
ABRS-P模型是利用以前建立的机器学习管道开发的,该管道被称为**聚类约束注意力多实例学习**(CLAM)。该管道可能旨在处理组织学切片的复杂性,并识别预测 ABRS 表达的模式。
训练数据集
该模型在癌症基因组图谱(TCGA)的多中心数据集上进行训练,其中包括接受手术切除的患者样本(n=336)。TCGA 是癌症基因组学的综合资源,为研究人员提供大规模数据集。
外部验证
随后在两个独立的数据集上对所开发的模型进行了验证:
- 手术切除系列(n=225)
- 活组织切片系列(样本数=157)
临床验证
在接受阿特珠单抗-贝伐单抗治疗的HCC患者多中心队列(122人)中进一步测试了该模型的预测价值。这些患者的活检样本用于评估模型预测真实世界治疗结果的能力。
患者群体
研究中使用的所有样本均来自成年患者(18 岁及以上)。
验证时间范围
验证集的取样时间跨度很长,从 2008 年 1 月 1 日到 2023 年 1 月 1 日,这表明对模型有效性的评估具有广泛性和纵向性。
主要目标
多中心验证的主要目的是确定与第一个活检系列的交叉验证中位分割阈值相比,ABRS-P 值的高低是否与开始治疗后的 PFS 相关。这是确定模型能否有效预测治疗反应的关键一步。
空间转录组学
该研究还采用了空间转录组学,这是一种可以对组织样本中的基因表达进行空间映射的技术。该技术用于将模型的**预测热图与组织样本的实际原位(在位)表达谱**关联起来。
总之,这项研究利用先进的机器学习技术开发并验证了一个模型,该模型可以根据组织学切片分析预测 HCC 患者的治疗反应。最终目标是利用该模型指导临床决策和个性化医疗,并有可能改善阿特珠单抗和贝伐珠单抗治疗 HCC 的疗效。广泛的验证过程,包括使用不同的样本类型和真实世界的临床数据,提高了 ABRS-P 模型的可信度和潜在的临床实用性。
1-3:Findings
该研究旨在开发和验证一种预测模型(ABRS-P),用于估计肝细胞癌(HCC)患者的阿特珠单抗-贝伐单抗反应特征(ABRS)。以下是要点总结和分析:
患者人口统计学
在抽样的 840 名患者中,男性占大多数(76%),女性占 24%。这一信息对于了解研究人群的人口统计学特征非常重要。
风险因素
该研究确定了 HCC 的常见风险因素,其中包括
- 酒精摄入量
- 乙型肝炎
- 丙型肝炎病毒感染
- 非酒精性脂肪性肝炎
这些风险因素在文献中均有详细记载,有助于了解样本人群的具体情况。
模型性能
在开发系列的交叉验证过程中,ABRS-P 值(模型预测值)与实际 ABRS 分数(ABRS 基因的平均表达量)之间的平均皮尔逊相关系数为 r=0.62。0.62 的相关系数表明,模型预测值与 ABRS 实际得分之间存在中等至较强的正相关关系。p 值始终低于 0.0001,表明观察到的相关性偶然发生的概率很低。
外部验证
ABRS-P 模型在外部验证系列中表现良好,在手术切除系列中的相关性为 r=0.60,在活检系列中的相关性为 r=0.53。这些结果表明,该模型在不同数据集和样本类型中具有稳健性和通用性。
临床结果
在接受阿特珠单抗-贝伐单抗治疗的 122 例患者的临床验证集中,对该模型预测患者预后的能力进行了测试。
与 "ABRS-P-低 "肿瘤患者(48人)相比,"ABRS-P-高 "肿瘤患者(74人)在开始治疗后的中位无进展生存期(PFS)明显更长。ABRS-P高肿瘤患者的无进展生存期为12个月,长于ABRS-P低肿瘤患者的7个月。
这一差异具有统计学意义(P=0.014),表明该模型的预测具有临床意义,有助于对患者进行**分层治疗**。
空间转录组学
空间转录组学分析表明,ABRS-P 值高的肿瘤区域 ABRS 评分明显更高,各种免疫效应因子上调。这一发现支持了模型预测的生物学合理性,并表明该模型正在识别更有可能对免疫疗法产生反应的肿瘤区域。
总之,研究结果表明,ABRS-P 模型是一种有效且可能对临床有用的工具,可用于**预测哪些 HCC 患者可能会对阿特珠单抗和贝伐珠单抗的治疗产生反应**。该模型在不同数据集上的表现、它与临床结果的关联以及来自空间转录组学的支持数据为进一步的研究和潜在的临床实践提供了坚实的基础,从而为 HCC 患者的个性化治疗决策提供指导。
1-4:Interpretation
研究报告的解释部分讨论了研究结果的意义和潜在影响。以下是对要点的总结和分析:
人工智能作为一种生物标志物
该研究表明,应用人工智能分析肝细胞癌(HCC)的数字切片可作为阿特珠单抗和贝伐珠单抗联合治疗患者无进展生存期(PFS)的生物标志物。这是人工智能在精准医疗领域的新应用,计算模型可以帮助预测患者对治疗的个体反应。
开发成本低廉的生物标志物
研究表明,与传统的生物标记物发现方法相比,基于人工智能的生物标记物可以以相对低廉的成本快速开发出来。这一点在肿瘤学领域尤为重要,因为及时、经济高效的诊断方法可对治疗决策和患者预后产生重大影响。
人工智能热图与空间转录组学的整合
该研究利用**人工智能热图(突出显示组织切片中具有某些预测特征的区域)**与空间转录组学相结合,提供了与治疗反应相关的分子特征的详细视图。这种综合方法可以加深人们对肿瘤对免疫疗法和抗血管生成疗法反应的生物机制的理解。
推广潜力
针对 HCC 开发的方法可应用于其他类型的癌症和疾病。这种交叉应用性是人工智能模型的一大优势,因为它们可以在不同的数据集上进行调整和训练,以应对各种医学挑战。
提高对治疗的理解
通过使用人工智能分析组织学切片和空间转录组学数据,该研究旨在提高对不同治疗方法如何在分子水平上发挥作用的理解。这种更深入的洞察力可以带来更好的治疗策略、确定新的药物靶点以及开发辅助诊断方法。
总之,这项研究的解释强调了人工智能在革新生物标记物发现和个性化医疗方面的潜力。研究结果表明,基于人工智能的模型可以预测 HCC 患者的治疗反应,为临床决策提供了一种前景广阔的工具。这种方法不仅适用于 HCC,而且有望在肿瘤学和其他疾病中得到更广泛的应用,从而为更个性化、更精确的医疗保健方法做出贡献。
二、Research in context
2-1:Evidence before this study
该内容介绍了在PubMed上进行的文献检索,以确定基于对整张切片图像的分析,使用人工智能(AI)预测患者对靶向疗法和免疫疗法敏感性的研究。该检索于2023年5月15日进行,使用的术语为 "人工智能 "或 "深度学习 "或 “机器学习”、"治疗 "和 “切片”,所收录的文章没有语言限制。
以下是对要点的总结和分析:
文献检索
研究人员对 PubMed 数据库进行了全面搜索,以收集相关研究。PubMed 是一个被广泛使用的生物医学文献资源,因此是此类研究的合适起点。
研究空白
搜索结果显示,利用人工智能从整张切片图像直接评估患者对靶向治疗和免疫治疗敏感性的研究相对较少。这表明该领域仍处于早期阶段,有很大的发展和探索空间。
创新方法
研究人员特别感兴趣的是,能否利用基于人工智能的病理学方法估算出与抗癌药物反应相关的基因特征。基因特征是一种基因表达模式,可用于预测患者对特定治疗的反应。
未开发领域
该内容指出,还没有研究调查过使用**基于人工智能的病理学来估计与药物反应相关的基因特征**。这表明,人工智能、病理学和基因特征的交叉是一个新领域,在推进精准医疗方面具有尚未开发的潜力。
总之,文献检索凸显了利用人工智能从整张切片图像预测治疗反应的新颖性。研究结果表明,开发基于人工智能的工具大有可为,这些工具可以分析组织学切片,深入了解表明患者对抗癌疗法潜在反应的基因特征。这将带来更加个性化的治疗策略,改善癌症患者的治疗效果。
2-2:Added value of this study
内容讨论了这项研究的附加值和贡献,涉及使用基于人工智能的病理学作为接受系统疗法的癌症患者的生物标志物。
以下是要点总结和分析:
基于人工智能的病理学作为生物标志物
该研究表明,人工智能可用于预测癌症患者的治疗反应。通过应用人工智能分析组织学切片,研究人员创建了一种潜在的生物标志物,可以指导阿特珠单抗和贝伐珠单抗等系统疗法的治疗决策。
人工智能模型的稳健性
这项研究的结果凸显了人工智能模型的稳健性,因为它是**在两个独立的数据集上验证**的,这两个数据集在临床和技术上与原始开发集存在差异。这种交叉验证对于确保模型的预测结果可靠并适用于不同的患者群体和成像条件至关重要。
深度学习与空间转录组学的整合
研究人员开发了一种将深度学习预测热图与空间转录组学数据相结合的管道。这种集成方法可以**将肿瘤微环境中与人工智能治疗反应预测相关的分子特征可视化**。
原位验证
该研究提供了原位验证,表明人工智能模型预测具有高阿替珠单抗-贝伐单抗反应特征(ABRS)的肿瘤区域与预测具有低ABRS值的区域相比,确实表现出更高的ABRS表达。人工智能预测与实际分子表达水平之间的这种相关性增加了模型准确性和生物学相关性的可信度。
总之,这项研究的附加值在于它证明了基于人工智能的病理学可以作为预测癌症患者治疗反应的可靠生物标志物。深度学习与空间转录组学的整合为了解治疗反应的分子基础提供了一个强大的工具,其详细程度是以前无法做到的。这种方法有可能应用于多种癌症和治疗策略,从而提供更加个性化和有效的治疗方案。
2-3:Implications of all the available evidence
内容讨论了研究结果的意义以及基于人工智能的生物标志物对临床实践和研究的潜在影响。以下是对要点的总结和分析:
前瞻性验证
研究中开发的基于人工智能的新型生物标记物需要前瞻性验证,以确认其临床实用性。前瞻性验证包括**在治疗前对一组新患者进行生物标志物测试,以确定它是否能准确预测治疗反应**。这是将生物标志物确立为临床决策工具的关键一步。
快速、低成本的替代方法
如果得到验证,与现有技术相比,这种基于人工智能的生物标志物可提供更快、更经济的选择,用于识别可能从特定治疗(如免疫疗法)中获益的患者。这对于精准医疗尤为重要,因为精准医疗的目标是为患者匹配最有可能对其有效的治疗方法。
将人工智能热图与空间剖析相结合
该研究将人工智能生成的热图与空间转录组学分析相结合的方法可扩展到其他类型的癌症和疾病。这种方法可以详细了解肿瘤微环境以及与治疗反应或耐药性相关的分子特征。
了解生物机制
人工智能与空间图谱分析的整合有可能加深我们对决定肿瘤如何响应或抵抗治疗的生物机制的理解。这些知识可以帮助我们发现新的治疗靶点,制定更有效的治疗策略。
总之,这项研究的意义在于,基于人工智能的生物标志物可以彻底改变我们识别和治疗癌症患者的方式。对治疗反应进行快速、低成本评估的潜力可以带来更个性化、更有效的治疗。此外,人工智能与空间转录组学的结合可能成为揭示肿瘤生物学复杂性的有力工具,并在各种疾病中得到广泛应用。不过,这种方法的实用性还需要通过前瞻性临床试验来证实,以确保它能在现实世界中造福患者。
三、Introduction
导言提供了有关肝细胞癌(HCC)的背景信息以及开展这项研究的理由。以下是对要点的总结和分析:
肝细胞癌(HCC)
HCC是一种常见的致命癌症,在全球总发病率中排名第六,在癌症相关死亡人数中排名第三。高死亡率凸显了有效治疗的必要性。
联合疗法
阿特珠单抗-贝伐单抗联合疗法是晚期 HCC 的标准治疗方法。
- 阿特珠单抗是一种**免疫检查点抑制剂**,可帮助免疫系统攻击癌细胞;
- 贝伐单抗是一种**抗血管生成药物**,可抑制肿瘤供血血管的生长。
有限的反应率
尽管有这种联合疗法,但**只有少数患者有反应,这凸显了开发生物标志物以指导治疗决策**的重要性。
阿替珠单抗-贝伐单抗反应特征(ABRS)
Zhu及其同事在接受阿特珠单抗-贝伐单抗治疗的患者中发现了一个与无进展生存期相关的基因特征(ABRS)。该特征中的基因**涉及 T 细胞活化和先天性免疫**,这对于癌症的免疫反应至关重要。
基因特征的挑战
虽然 ABRS 等基因特征具有作为生物标记物的潜力,但其实际应用受到评估所需的高成本和专业技术的限制。标准化问题也影响了它们的可靠性,这就是为什么除了在乳腺癌中的某些应用外,它们在临床实践中没有得到广泛应用的原因。
组织切片的可获得性
组织切片可随时从病理部门获得,可通过人工智能处理来预测分子改变。这种方法已被用于预测癌症中几种相关的分子改变。
人工智能在预测治疗反应中的应用
利用人工智能预测基于组织学切片的靶向治疗和免疫治疗反应的研究较少,尤其是在 HCC 中。本研究旨在探索基于人工智能的组织学分析是否可作为晚期 HCC 全身治疗的生物标记。
训练人工智能模型
由于来自接受治疗的HCC患者的组织学样本有限,直接根据临床结果训练人工智能模型具有挑战性。作为一种变通方法,研究人员选择创建一种预测 ABRS 表达的算法,将其作为替代生物标志物。
总之,导言为研究奠定了基础,强调了需要生物标志物来提高阿特珠单抗-贝伐单抗治疗 HCC 的有效性。研究人员建议使用人工智能分析组织学切片,作为预测治疗反应的潜在解决方案,以规避传统基因特征生物标志物的局限性。这项研究的方法可以为HCC患者带来更个性化、更有效的治疗策略。
四、Methods
4-1:Study design and participants
研究设计和参与人员部分详细介绍了用于收集和分析研究数据的方法。以下是对要点的总结和分析:
研究工作流程
研究的工作流程如图 1 所示,在此不做介绍,但可以描述数据收集、模型训练和验证的步骤。
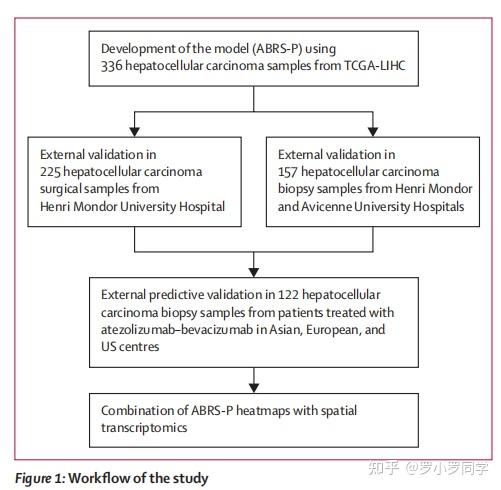
患者队列
对于所有患者队列,每个患者使用一个组织学样本。这确保了用于模型训练和测试的数据的一致性。
TCGA-LIHC 数据集
癌症基因组图谱肝肝细胞癌(TCGA-LIHC)公共数据集用于训练模型。该数据集包括在美国 20 多个中心接受手术切除的原发性 HCC 患者。
纳入标准
由两名肝脏病理学家明确诊断为HCC的患者、有数字化组织学切片的患者和有基因表达谱数据的患者均被纳入。
外部验证系列
两个外部验证系列用于测试模型的预测能力:
- 法国亨利-蒙多大学医院的手术切除系列(n=225)。
- 法国两家肝脏中心的活检系列(n=157)。
验证集的差异
这些验证集在取样方法、染色方案和 RNA 测序技术方面与 TCGA-LIHC 系列不同。
第四队列
第四批接受肝活检的HCC患者被用来检验模型对阿特珠单抗-贝伐单抗治疗结果的预测价值。该队列包括**来自亚洲、欧洲和美国 20 个临床中心的 122 名患者**。
患者特征
数据集包括不同来源的 HCC 病例,包括与乙型肝炎病毒(HBV)、丙型肝炎病毒(HCV)和酒精有关的病例。性别数据取自医疗记录,但种族和民族数据仅在 TCGA-LIHC 系列中可用。
时间范围
研究在 2021 年 10 月 1 日至 2023 年 8 月 20 日期间进行。
总之,该研究的设计非常严谨,涉及多个患者队列和外部验证集,以确保模型预测的稳健性和可推广性。TCGA-LIHC数据集的使用和不同患者特征的纳入增强了研究的临床相关性。伦理方面的考虑确保了该研究在患者同意和监督方面符合高标准。该研究的时间安排表明,在相对较长的时间内,为收集和分析数据付出了巨大的努力。
4-2:RNA sequencing and spatial transcriptomics
内容讨论了研究中使用的 RNA 测序和空间转录组学方法。以下是对要点的总结和分析:
RNA测序
对于前两个验证系列的批量 RNA 测序,从样本中对肿瘤区域进行大体解剖并提取 RNA。这一过程包括将肿瘤细胞从周围组织中分离出来,以确保测序数据能反映癌细胞的基因构成。
文库制备
使用 Lexogen QuantSeq-FWD 3’mRNAASeq 试剂盒制备文库,该试剂盒专为高通量 mRNA 转录本测序而设计。mRNA 是蛋白质合成的主要分子,通常也是治疗干预的目标。
测序平台
测序是在 Illumina NovaSeq 6000 仪器上进行的,这是一种高通量测序平台,以速度快、准确性高而著称。NovaSeq 的使用表明,研究人员能够在相对较短的时间内处理大量样本。
空间转录组学
为了进行空间转录组学研究,研究人员**从手术切除验证集中随机选择了四个肿瘤**。肿瘤切片被放置在FFPE Visium空间基因表达载玻片中,该载玻片可捕捉并保存组织样本中的空间信息。
切片选择
肿瘤的随机选择是使用 Microsoft Excel 中的 ALEA 函数完成的,这是一个简单的随机数生成器。这可确保选择无偏见,并能代表整个验证集。
测序仪器
空间转录组文库在另一个 Illumina 平台 NextSeq 2000 仪器上测序。使用 100 个循环的流动池表明这是一种高深度测序方法,可提供对转录组的详细了解。
基因表达分析管道
附录中报告了基因表达分析管道的详细规程,其中包括数据处理、归一化和统计分析等步骤,以解读测序数据。
总之,本研究利用最先进的 RNA 测序和空间转录组学技术,从肿瘤样本中获取了全面的基因组数据。这些方法对于了解肿瘤行为的分子基础以及确定潜在的生物标记物和治疗靶点至关重要。高通量测序平台的使用和样本的随机选择增强了研究结果的可靠性和可推广性。
4-3:Image preprocessing and deep learning networks
内容讨论了研究中用于分析肝细胞癌(HCC)组织学图像的图像预处理和深度学习网络,目的是预测阿特珠单抗-贝伐单抗反应特征(ABRS)。以下是要点总结和分析:
图像预处理
组织学图像中的组织区域是通过聚类约束注意力多实例学习(CLAM)管道检测到的。该管道旨在识别和分割图像中的肿瘤区域,这是准确分析的关键步骤。
注释
病理专家(JC)使用**QuPath软件**对肿瘤区域的多边形感兴趣区进行标注。该软件通常用于数字病理学,可在图像中精确标注感兴趣区。
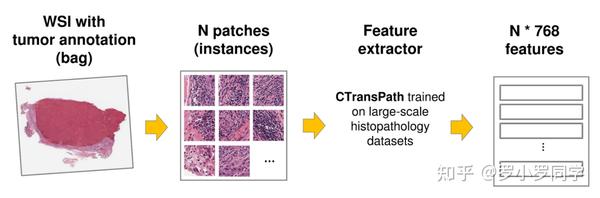
生成补丁
注释的肿瘤区域和 CLAM 管道检测到的区域的交叉区域被分割成**256×256 像素的补丁,没有重叠。这一步对于创建可管理的数据集进行深度学习分析非常重要,并可确保每个补丁代表肿瘤的一个不同区域**。
深度学习网络
研究中使用的深度学习网络基于 CLAM 架构,但针对回归分析进行了修改,涉及预测连续数值。
该网络使用无监督对比学习转换器(CTransPath)将每个补丁编码为 768 维的特征嵌入**。然后将该嵌入缩减为 512 维,并为每个补丁学习注意力分数。使用**Softplus 激活函数的全连接层根据 512 维特征为每个补丁预测一个非负连续值。
数据表示
使用基于变换器的方法对补丁进行编码,表明重点在于捕捉图像中的局部和全局特征,这对预测 ABRS 表达等任务很有帮助。
代码可用性
用于深度学习的代码可在线获取,这种做法提高了研究的透明度和可重复性。
总之,本研究采用了复杂的图像处理和深度学习技术来分析 HCC 的组织学图像。CLAM 的使用和对回归分析的修改表明,该研究非常注重识别可预测治疗反应的特定分子改变。代码的在线提供进一步提高了研究结果的可信度,并允许其他研究人员复制这些方法。
4-4:Network training, performance evaluation, and model interpretability
内容讨论了该研究的网络训练、性能评估和模型可解释性等方面。以下是对要点的总结和分析:
模型训练
人工智能模型的训练目的是**使其预测值与实际 ABRS 分数之间的均方误差最小**。ABRS 评分来自一组与治疗反应相关的基因的表达水平。
训练策略
使用**十次蒙特卡洛交叉验证**策略进行训练。这包括将数据集分成十个不同的子集,在九个子集上训练模型,然后在剩下的一个子集上进行验证。该过程重复十次,每次迭代使用不同的子集进行验证。
数据分割
在开发集(TCGA-LIHC)中,患者被随机分为**训练集(60%)、验证集(20%)和测试集(20%)**。这样可以确保模型在不同的数据集上进行训练,并在未见过的数据上进行测试,以评估其泛化能力。
性能评估
使用皮尔逊相关系数 ®、95% 置信区间 (CI) 和 p 值对模型的性能进行评估。皮尔逊相关性评估预测 ABRS 分数与真实 ABRS 分数之间的线性关系,数值越高,表示相关性越强,预测效果越好。
集合模型
对于前两个外部验证集,来自十次交叉验证的预测值通过**平均池**进行了组合,以减少任何单一验证集的影响,并降低误导结果的风险。
分层
根据常见的**临床特征(年龄、性别、疾病分期)和病理特征**(肿瘤大小、血管侵犯、分化程度)进行分层后,还对模型的性能进行了评估。这有助于了解模型在不同患者亚群中的表现,这对临床应用非常重要。
治疗结果分类
对于接受阿特珠单抗-贝伐单抗治疗的患者系列,根据第一次活检系列的中位分裂阈值将预测结果分为 ABRS-P 高或 ABRS-P 低类别。最终分类采用多数表决法,在五次或五次以上迭代中预测为 ABRS-P-高的样本被归类为 ABRS-P-高。
模型可解释性
为提高模型的可解释性,将斑块预测值重新调整为(0,1)范围,并通过平均集合进行组合。利用每个斑块的空间坐标绘制了彩色地图,以说明最终预测结果。这种可视化表达方式有助于了解模型在图像中的哪个位置做出了预测,以及这些预测的可信度如何。
总之,该研究采用了严格的方法来训练和评估人工智能模型,使用交叉验证和各种性能指标来确保其可靠性。集合模型和分层分析有助于验证模型的稳健性和临床相关性。色彩图等模型可解释性技术让人们深入了解模型是如何处理和解释图像数据的,这对理解其决策过程至关重要。
4-5:Outcomes and statistical analysis
内容介绍了研究中使用的结果和统计分析,以评估人工智能预测的 ABRS-P(阿替珠单抗-贝伐珠单抗反应特征)与无进展生存期(PFS)和总生存期(OS)等患者预后之间的关联。以下是要点总结和分析:
主要终点
主要终点是**ABRS-P与无进展生存期的关系**,无进展生存期是指从阿特珠单抗-贝伐单抗治疗开始到疾病进展或死亡的时间。未进展或死亡的患者以其最后一次可评估的肿瘤评估日期为截止日期。
补充终点
总生存期定义为从开始接受系统治疗到死亡或最后一次随访的时间。未死亡的患者在其最后一次随访日期被剔除。
生存曲线比较
使用 Kaplan-Meier 法和对数秩统计法比较 ABRS-P 高样本患者与 ABRS-P 低样本患者的生存曲线。
单变量和多变量分析
使用 Cox 比例危险度回归模型对 PFS 进行单变量分析。为避免偏差,单变量分析中的所有变量均纳入多变量分析。
皮尔逊相关性
计算皮尔逊相关系数以评估 ABRS-P 与实际 ABRS 评分之间的线性关系。
空间转录组学分析
对于空间转录组学,使用 **Mann-Whitney U 检验**来比较每个样本中 ABRS-P 值最高和最低的前 100 个斑块的 ABRS 分数。使用 Seurat 中的 FindMarkers 功能和 MAST 测试参数确定转录组中的差异表达基因。
统计意义
所有测试均为双尾测试,P 值小于 0.05 即为具有统计学意义。
总之,该研究结合使用了生存分析、相关性和转录组学来验证人工智能预测的 ABRS-P 与患者预后之间的关联。Kaplan-Meier 分析提供了生存曲线的直观表示,而 Cox 回归模型则评估了各种临床和病理变量对生存的影响。皮尔逊相关性评估了人工智能模型预测的准确性,而空间转录组学分析则提供了人工智能预测的分子基础。统计分析可确保结果的稳健性,并确保观察到的任何关联都具有统计学意义。
五、Results
内容概述了这项研究的方法,重点介绍了用于预测肝细胞癌(HCC)患者阿特珠单抗-贝伐单抗反应特征(ABRS)的ABRS-P模型的训练和验证。以下是要点总结和分析:
研究人群
该模型使用公开的 TCGA-LIHC 数据集进行训练,其中包括 336 名接受手术切除的 HCC 患者。大多数患者为男性(68%),中位年龄为 61 岁。HCC最常见的风险因素是饮酒。
模型架构
ABRS-P 模型使用的深度学习架构改编自之前发布的管道,并进行了修改以执行回归分析。这表明**其重点在于数值预测而非分类**。
训练和性能
使用交叉验证对模型进行训练,最佳模型的皮尔逊相关系数(r)为 0.71,集合模型的皮尔逊相关系数(r)为 0.86。这表明该模型的预测结果与 ABRS 实际得分之间存在很强的线性关系。
外部验证
该模型的性能在两个独立的 HCC 患者系列中进行了测试。这些外部数据集在染色方案、组织学取样、基因表达谱分析技术和切片编码格式方面与开发集不同,这对模型的通用性非常重要。
总之,本研究的方法论展示了使用大型公共数据集对 ABRS-P 模型进行的训练和初步验证。使用皮尔逊相关性对模型的性能进行了评估,结果表明该模型具有很强的预测能力。交叉验证的使用增强了结果的可靠性。在具有不同特征的数据集上进行外部验证是确保模型能够泛化到真实世界场景和不同实验条件的关键步骤。
内容详细介绍了研究中使用的**首个验证数据集,**以评估 ABRS-P 模型的性能。以下是对要点的总结和分析:
验证数据集特征
第一个验证数据集包括在亨利-蒙多大学医院接受手术切除的 225 名患者。大部分患者为男性(80%),中位年龄为 64 岁。
临床和病理特征
患者被确诊为 HCC,具有不同的临床、生物学和病理学特征。最常见的危险因素是 HBV 感染和酒精摄入。相当一部分患者具有侵袭性形态特征,如分化不良、大血管侵犯、微血管侵犯和卫星结节。
ABRS-P模型性能
该数据集中患者的切片使用与开发集相同的管道进行处理。ABRS-P 值与实际 ABRS 评分仍有显著相关性,皮尔逊系数为 0.60 (95% CI 0.51-0.68; p<0.0001)。这一结果表明,该模型的预测与这一独立数据集中的实际 ABRS 评分一致,验证了该模型的性能。
总之,第一个验证数据集表明,尽管染色方案、组织学取样、基因表达谱分析技术和切片编码格式与开发集不同,ABRS-P 模型仍能准确预测各类 HCC 患者的 ABRS 评分。这是验证该模型通用性和临床应用潜力的重要一步。
内容讨论了使用纯活检数据集验证 ABRS-P 模型及其与接受阿特珠单抗-贝伐单抗治疗的肝细胞癌 (HCC) 患者无进展生存期的关系。以下是要点总结和分析:
活检数据集
157名患者的活检数据集用于验证ABRS-P模型。与第一个验证数据集相比,该数据集的性别分布相似,但中位年龄略高。
ABRS-P 模型的性能
ABRS-P 模型在该数据集中的表现显示皮尔逊相关系数为 0.53,表明与第一个验证数据集相比表现略有下降。不过,相关性仍然显著,表明尽管样本类型不同,模型仍然具有预测性。
性能分层
根据常见的临床和病理特征进行分层后,ABRS-P 模型仍具有预测价值,但对 60 岁或以下患者和肝硬化患者的预测效果明显下降。
与无进展生存期的关系
ABRS-P 模型随后在接受阿特珠单抗-贝伐单抗治疗的 122 例患者队列中进行了评估。与 ABRS-P 低的肿瘤患者相比,ABRS-P 高的肿瘤患者的中位无进展生存期明显更长(12 个月 vs 7 个月;P=0.014)。这种关联在多变量分析中仍具有重要意义。
总生存期
ABRS-P 模型预测结果与总生存期无关,这表明虽然该模型可预测治疗反应,但可能不是总预后的有力指标。
总之,纯活检数据集验证了 ABRS-P 模型的性能,但与手术切除数据集相比,准确性略有降低。ABRS-P高的肿瘤与更长的无进展生存期之间的明显关联支持该模型作为预测HCC患者治疗结果的生物标记物的潜在用途。该模型与总生存期缺乏关联,因此需要进一步研究,以了解该模型的局限性和在临床实践中的潜在应用。
内容讨论了如何利用空间转录组学深入了解手术验证系列中四个肝细胞癌(HCC)样本中与 ABRS-P 值高低相关的基因表达谱。以下是要点总结和分析:
空间转录组学
对四个 HCC 样本进行了空间转录组学研究,以便将基因表达模式与 ABRS-P 模型的预测结果关联起来。这种方法可**对每个肿瘤中 ABRS-P 值最高和最低的前 100 个斑块进行比较**。
ABRS 评分比较
在四个病例中的三个病例中,发现 ABRS-P 值高的肿瘤区域的 ABRS 评分明显高于 ABRS-P 值低的区域。这表明该模型可识别出具有独特分子特征的区域,而这种特征可预测 ABRS 评分。
相关性分析
在这些斑块中,ABRS 评分与 ABRS-P 预测值之间的相关性显示出较低的皮尔逊系数(0.14-0.37),但在四个案例中有三个案例的 P 值显著,这表明在这些斑块中,实际 ABRS 评分与模型预测值之间存在微弱但具有统计学意义的关系。
差异基因表达
对整个转录组的分析表明,在所有四个案例中,ABRS-P 高的斑块和 ABRS-P 低的斑块之间的基因表达存在显著差异。这些差异表明,该模型正在识别具有不同免疫和细胞特征的区域。
免疫概况特征
在不同的样本中,ABRS-P-高斑块中的各种免疫和细胞标志物上调,包括 T 细胞标志物、肝细胞标志物、参与炎症、干扰素信号转导、抗原递呈和细胞外基质产生的基因。
总之,空间转录组学分析提供的证据表明,ABRS-P 模型正在识别具有特定免疫和分子特征的肿瘤区域。这些发现支持了该模型预测的生物学合理性,并表明该模型正在捕捉与 HCC 患者治疗反应相关的生物学特征。这些结果凸显了将人工智能与分子图谱技术相结合,以促进对肿瘤生物学的了解并为治疗决策提供依据的潜力。
六、Discussion
6-1:研究结果的意义和影响
研究报告的讨论部分深入探讨了研究结果的意义和影响。以下是对要点的总结和分析:
人工智能作为一种生物标志物
该研究表明,将人工智能应用于肝细胞癌(HCC)的数字化组织学切片,可作为阿特珠单抗-贝伐单抗治疗患者无进展生存期(PFS)的生物标志物。这一发现意义重大,因为它表明人工智能有助于预测像 HCC 这样的异质性疾病的治疗结果。
关注基因表达
研究人员选择关注基因表达(ABRS)而非风险因素或遗传缺陷,因为之前的研究并未发现这些因素对接受阿特珠单抗-贝伐珠单抗治疗的患者的临床结果有任何影响。
复杂性和成本
虽然已经测试了几种预测治疗结果的基因特征,但由于其复杂性和成本问题,大多数基因特征尚未在临床实践中应用。人工智能方法旨在克服这些障碍。
弱监督学习
该研究是首批利用弱监督学习从病理数据中预测基因特征表达的连续生物标志物的研究之一。这意味着人工智能模型是在有限的标注数据下进行训练的,依靠其他信息来源来指导其学习。
空间转录组学
这项研究也是首次利用空间转录组学对预测结果进行原位验证,空间转录组学提供了详细的肿瘤分子图谱。
模型的鲁棒性
使用不同的染色方案、编码格式和表达谱分析技术,对来自不同中心的样本进行了模型稳健性测试。这种广泛的外部验证提高了模型的可信度,并表明它可以推广到各种临床环境中。
临床适用性
使用固定阈值将样本划分为 ABRS-P 高或 ABRS-P 低,对于证明该生物标记物可用于临床实践至关重要。
概括性
该模型的预测对具有不同风险因素(如饮酒和 HBV 感染)的患者具有很好的普适性,这表明它可以处理不同的患者群体。
模型透明度
该研究旨在通过结合人工智能热图和空间转录组学来解决人工智能模型缺乏透明度的问题。这种方法使研究人员得以证实,在某些情况下,ABRS-P 值较高的肿瘤区域会过度表达 ABRS 和其他免疫效应因子。
未来的信心和信任
虽然分析并不能完全解释模型,但它有助于确定影响预测的分子特征。这一工作流程可能有助于建立对基于人工智能的病理生物标志物的信心和信任,以便将来在临床实践中采用。
总之,本研究的讨论强调了人工智能作为预测 HCC 患者治疗结果的生物标志物的潜力。广泛的外部验证、对不同患者群体的推广以及空间转录组学的整合为该模型的稳健性和潜在临床实用性提供了证据。通过深入了解影响人工智能预测的分子特征,该研究还解决了人们对模型透明度的担忧。
6-2:研究的局限性及其对未来研究的潜在影响
内容讨论了研究的局限性及其对未来研究的潜在影响。以下是对要点的总结和分析:
研究局限性
- 回顾性研究: 这项研究是回顾性的,这意味着它回顾了现有的数据。这可能导致数据缺失或不完整,前瞻性验证对于在临床应用前确认模型的预测能力至关重要。
- 前瞻性验证: 未来的研究需要对模型进行前瞻性验证,即在治疗前进行验证,以确保其临床实用性。
- 人群代表性: 应在研究未包括的人群中调查该模型的性能,如非洲人群和更多的亚洲样本,以确保其适用于不同人群。
- 其他疗法: 为完全排除阿特珠单抗-贝伐单抗治疗的非特异性预后价值,该模型应在接受其他靶向治疗的患者中进行测试。
- 生存期指标: 将无进展生存期作为晚期HCC患者总生存期的替代指标受到了质疑,更长时间的随访可能会发现总生存期的差异。
- RNA碎片: 福尔马林固定石蜡包埋(FFPE)样本中的 RNA 分子是破碎的,这会限制空间转录组学对基线表达量极低的基因的检测。
- 空间转录组学的局限性: 空间转录组学并不能提供完全的可解释性,进一步研究可解释人工智能(XAI)对于更好地理解模型的行为和发现潜在缺陷非常重要。
未来研究与影响
- 前瞻性验证: 前瞻性验证对于确认模型的预测能力和临床实用性至关重要,然后才能考虑将其用于常规用途。
- 人群多样性: 研究模型在不同人群中的表现将确保其广泛的适用性。
- 多种疗法: 用其他疗法测试该模型将有助于确定其预测价值是专门针对阿特珠单抗-贝伐单抗的,还是更广泛适用的。
- 生存结果: 更长期的随访研究可能会发现总生存期的差异,而这些差异是单纯的无进展生存期所无法反映的。
- XAI研究: 可解释的人工智能研究有助于理解模型的行为并提高其透明度,这对临床应用至关重要。
总之,这项研究展示了基于人工智能的生物标记物在快速识别可能从特定治疗中获益的患者方面的潜力。将预测热图与原位基因表达谱分析相结合,可以深入了解肿瘤区域的分子特征,具有很高的预测价值。虽然这项研究还存在需要进一步研究的局限性,但其研究结果表明,这种方法可以应用于其他癌症或疾病,以加深对驱动治疗结果和反应的生物机制的理解。
七、补充文档分析
7-1:RNA sequencing
内容详细介绍了该研究在前两个验证系列(切除术和肝活检)中使用的 RNA 测序方法。以下是对要点的总结和分析:
RNA 提取
对于前两个验证系列中的每个病例,都从 5 毫米厚的切片中或直接从福尔马林固定石蜡包埋(FFPE)组织块中对肿瘤区域进行宏观解剖。在 Promega Maxwell 仪器上使用 Maxwell® RSC RNA FFPE Kit 提取 RNA,确保总 RNA 的质量、纯度和完整性。
RNA 质量控制
在安捷伦 2100 生物分析仪(Agilent 2100Bioanalyzer device)上使用 Pico 检测法对总 RNA 进行电泳监测,以确保其质量、纯度和完整性。
文库制备
使用QuantSeq FWD 3’mRNA Seq LEXOGEN试剂盒从不同数量的总RNA(从50到150纳克不等)中制备RNA测序文库。其中包括使用寡聚 dT 引物合成双链 cDNA,并进行 21 至 25 个循环的 PCR 扩增。
文库定量和测序
使用 KAPA 文库定量试剂盒对扩增和条形码文库进行汇集和定量。测序在 Illumina Novaseq 6000 仪器上以单线程模式(SR100)进行,目标是每个样本平均有 1000 万个读数。
数据管理与分析
数据管理、质量控制和初步分析由著名癌症研究机构居里研究所的生物信息学平台负责。
总之,该研究采用了严格的标准化流程进行 RNA 提取、文库制备和测序,以确保转录组数据的质量和可靠性。针对 FFPE 样品使用专门的试剂盒和仪器,凸显了研究人员对处理福尔马林固定组织的特殊挑战的关注。居里研究所的生物信息学平台确保由该领域的专家进行数据分析,从而提高了研究结果的可靠性。
7-2:Spatial transcriptomics
内容讨论了研究中用于分析肝细胞癌(HCC)肿瘤样本中基因表达空间分布的**空间转录组学方法**。以下是要点总结和分析:
RNA质量筛选
使用安捷伦 TapeStation 系统对样本进行 RNA 质量筛查,该系统根据制造商的建议检查 RNA 的完整性(DV200 分数 > 50%)。
空间基因表达载玻片和试剂
根据制造商的说明使用 Visium 空间基因表达载玻片和试剂盒。这些载玻片旨在捕捉和保存组织样本中的空间信息。
组织切片和捕获
从 FFPE 肿瘤块上切取 5 毫米厚的组织切片,并将其置于 FFPE Visium 空间基因表达载玻片的靶标框内。每个捕获区域包含约 5,000 个基因表达点,这些点是捕获和分析 RNA 的特定区域。
空间条形码和探针杂交
载玻片上的斑点包括一个部分读取 1 测序引物、一个 16-nt 空间条形码、一个 12-nt 唯一分子标识符(UMI)和一个 30-nt poly(dT) 序列。这些成分可以对捕获的 RNA 进行空间和分子鉴定。
玻片处理
玻片去石蜡、苏木精-伊红染色、盖玻片并以 x40 倍率扫描。然后取下盖玻片,进行脱交联步骤,以释放组织中的 RNA。
库制备
释放的 RNA 与探针杂交,连接形成连接产物,然后从组织切片中释放出来。连接产物通过添加 UMI、部分读数 1 和空间条形码进行扩展,用于制备测序文库。
测序
使用 Illumina San Diego 系统,在带有 P3 流动池(100 个循环)的 NextSeq 2000 仪器上对制备的文库进行测序。
总之,该研究中使用的空间转录组学方法提供了肿瘤空间基因表达的详细分子图谱,可**识别肿瘤内具有明显分子特征的特定区域。这项技术尤其有助于了解肿瘤微环境中复杂的相互作用,并确定潜在的生物标记物和治疗靶点**。专用设备和试剂的使用可确保所获数据的质量和准确性。
7-3:Gene expression analysis pipeline
内容介绍了研究中使用的基因表达分析管道,包括基于测序的方法和空间转录组学方法。以下是对要点的总结和分析:
测序数据转换
- 使用 DESeq 软件包中的 varianceStabilizingTransformation 函数对测序得到的原始数据进行转换。
- 这种转换基于从拟合的离散-均值关系中计算出的大小因子,有助于对数据进行归一化处理,并考虑到文库大小的差异。
- ABRS 分数根据特征基因的算术平均值计算,如果 KLRC3 未在所有样本中检测到,则将其排除在外。
空间转录组学数据处理
- 使用 Space Ranger 1.3.1 将 Illumina 测序仪的碱基调用文件(BCL)解复用为 FASTQ 文件。
- 使用 Space Ranger 1.3.1 和 10X genomics 提供的 Visium 人类转录组探针集 v1.0 GRCh38-2020-A 分析这些 FASTQ 文件和 tiff 格式的对齐组织学图像。
- 使用的探针集不包括伪基因 CXCR2P1,因此只分析了 10 个 ABRS 标志基因中的 8 个。
- 每个点测得的中位基因平均数为 2,882 个,范围在 2,426 至 3,794 个之间。
- 使用 Space Ranger 对数据进行预处理后,通过 Seurat 4.3.0 输入到 R 中,其中过滤掉了少于 300 个基因的空间点和计数少于 5 个空间点的基因。
- 使用 Seurat 函数 SCTransform(带 v2 正则化和 "assay=spatial "参数)对每个样本的原始计数进行归一化处理。
总之,基因表达分析管道是一个多步骤过程,涉及测序数据转换和空间转录组学数据处理。DESeq 转换是标准化测序数据的常用方法,而 Space Ranger 和 Seurat 工具则用于分析和预处理空间转录组学数据。过滤步骤确保了只有相关且代表性强的数据才会被用于进一步分析,从而提高了研究结果的稳健性。
7-4:Images preprocessing
内容详细介绍了研究中针对不同数据集使用的图像预处理步骤。下面是对要点的总结和分析:
TCGA-LIHC 发现系列
- 使用 Aperio 扫描仪对WSI进行数字化处理,放大倍数为 ×20(每像素 0.50 微米,MPP)或 ×40(每像素 0.25 微米,MPP)。
- 从 ×40 切片中提取 512 x 512 像素的图像片段,并进行降频处理,使其与 ×20 切片的分辨率和 256 x 256 像素的模型输入尺寸相匹配。
前两个外部验证系列
- WSI使用 Hamamatsu(ndpi 格式)或 Aperio 扫描仪(svs 格式)以 ×20 或 ×40 倍率进行数字化。
- 对于 ×20 WSI(ndpi:0.46 MPP,svs:0.50 MPP),提取的图像块为 256 x 256 像素的正方形大小。
- 对于 ×40 ndpi WSI(0.23 MPP),从第一个降采样级别(降采样系数 = 2)提取的图像补丁大小为 256 x 256 像素。
- 对于 ×40 svs WSI(0.25 MPP),从 512 x 512 像素大小的最高分辨率(无下采样级别,下采样系数 = 2)中提取图像补丁,然后下拉取样至 256 x 256 像素。
免疫治疗患者系列
- WSI以各种格式(mrxs、svs、ndpi、vsi、qptiff、tiff 和 tif)扫描,放大倍数各不相同(×20:0.24 MPP,×40:0.19 MPP;×20:0.50 MPP,×40:0.25 MPP;×40:0.23 MPP;×40:0.14 MPP)。
- 与之前的系列相似,对于放大倍数为[0.46,0.50]的WSI,提取的图像片段大小为 256 x 256 像素的正方形。
- 对于放大倍数为[0.23, 0.25]的WSI,如果存在降低取样系数为 2 的降低取样级别,则从降低取样级别提取图像补丁,否则从最高分辨率提取图像补丁,并降低取样系数为 2。
- 对于 ×40 mrxs WSI,从最高分辨率中提取图像片段,并按 2.5 的系数降低取样。
- 使用 BfConvert 命令行工具将 ×40 vsiWSI转换为**金字塔型** ome.tif 格式,并从系数为 4 的下采样水平提取图像补丁。
总之,图像预处理步骤包括从数字化组织学切片中提取不同放大倍率和分辨率的图像补丁。然后,对这些图像片段进行降采样,使其大小一致,以符合模型输入要求。图像数据的这种标准化对于准确的深度学习分析至关重要,可确保模型能够处理来自不同来源、具有不同特征的图像。使用不同的扫描格式以及对每种格式的特定处理步骤的需求,凸显了处理不同数据集的复杂性。
7-5:Deep learning networks
内容讨论了研究中用于分析肝细胞癌(HCC)组织学图像的深度学习网络,目的是预测阿特珠单抗-贝伐单抗反应特征(ABRS)。以下是要点总结和分析:
组织区域检测
使用聚类约束注意力多实例学习(CLAM)管道检测组织区域。该管道旨在识别和分割图像中的肿瘤区域,这是准确分析的关键步骤。
注释和补丁生成
肿瘤区域由病理专家(JC)使用 QuPath 软件注释为多边形感兴趣区。注释的肿瘤区域和 CLAM 管道检测到的区域的交叉区域被分割成 256x256 像素的无重叠斑块,分辨率约为 0.5 µm/像素。
深度学习网络架构
深度学习网络基于 CLAM 架构,但针对回归分析进行了修改。根据**多实例学习**(MIL)假设,每个斑块被视为一个实例,整个WSI被视为一个包。
特征嵌入
在将 WSI 加载到 GPU 内存时,使用无监督对比学习转换器(CTransPath)将每个补丁编码为 768 维的特征嵌入。
注意力网络
通过一个可训练的全连接(FC)层,将每个补丁的特征嵌入进一步缩减到 512 维。注意力网络由两个平行的 FC 层组成,具有不同的激活函数(Tanh 和 Sigmoid)。两个层的输出相乘,为每个贴片学习注意力分数。
补丁预测
使用具有 Softplus 激活功能的 FC 层,根据 512 维特征预测每个补丁的非负连续值。
预测聚合
对 N 个贴片的注意力分数进行Softmax处理,并将其作为权重,将贴片预测结果汇总为 WSI 预测结果。
示意图
架构示意图如下:
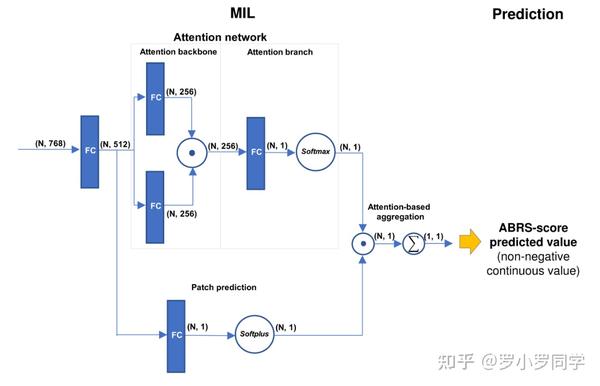
总之,本研究采用了复杂的深度学习网络架构来分析 HCC 的组织学图像,以预测 ABRS 的表达。使用 CLAM 管道进行组织区域检测和使用 MIL 方法处理 WSI 数据是该方法的显著特点。注意力网络增加了复杂性,可提高模型捕捉图像相关特征的能力。代码的在线提供提高了研究的透明度和可重复性。
7-6:Network training and performance evaluation
内容讨论了研究中使用的人工智能模型的网络训练和性能评估。以下是对要点的总结和分析:
模型训练
使用均方误差(MSE)损失函数对模型进行训练,以最小化 ABRS 预测分数与真实分数之间的差异。
Adam 优化器的学习率为 2e-4,L2 权重衰减为 1e-5。
训练至少持续 50 个时程,如果验证损失连续 20 个时程没有减少,则提前停止训练。使用最佳历时的模型。
采用十次蒙特卡罗交叉验证策略,将开发序列中的患者随机分为训练集(60%)、验证集(20%)和测试集(20%)。
性能评估
使用皮尔逊相关系数、95% 置信区间(CI)及其 p 值评估模型的性能。
对于前两个外部验证序列,十次交叉验证的预测结果取平均值,以减少任何单一验证集的影响。
使用相同的方法(皮尔逊系数)对常见的临床和病理特征进行分层后,也对模型的性能进行了评估。
对于来自接受治疗的患者的样本,预测结果使用第一个活检系列的中位分裂阈值分为 ABRS-P 高或低。使用多数票表决确定最终分类,并使用对数秩检验比较两类患者的无进展生存期(PFS)。
训练和验证策略
蒙特卡洛交叉验证的使用确保了模型在不同的数据集上得到训练,并在未见过的数据上得到验证,以评估其泛化能力。
- 早期停止机制有助于防止过度拟合,并确保模型经过适当数量的历时训练。
- 根据临床和病理特征对验证集和测试集进行分层,可以更全面地评估模型在不同患者亚群中的表现。
总之,该研究采用了严格的方法来训练和评估人工智能模型,使用交叉验证和各种性能指标来确保其可靠性。早期停止和分层分析的使用有助于验证模型的稳健性和临床相关性。盲法分析进一步提高了结果的可信度。
7-7:Model interpretability
内容讨论了研究中使用的模型可解释性方法,以深入了解人工智能模型如何处理和解释图像数据。以下是对要点的总结和分析:
补丁预测重新缩放
补丁预测被重新缩放至 [0, 1] 范围,其中 1 代表最高预测值,0 代表最低预测值。
这一重新缩放步骤可确保预测结果采用适合解释的标准格式。
平均池化
通过平均汇集法,对所有十次交叉验证的重定标预测结果进行汇集。这个平均化过程有助于平滑预测中的任何波动,并提供一个更稳定、更有代表性的模型性能视图。
创建热图
每个斑块的空间坐标被用来创建彩色地图,红色代表预测值为 1,蓝色代表预测值为 0。
热图直观地显示了模型对每个斑块的预测,表明肿瘤的哪些区域可能与阿特珠单抗-贝伐单抗的反应相关。
临床研究意义
这些热图为临床研究提供了线索,突出了模型与治疗反应可能性较高相关的肿瘤区域。
这可以指导进一步研究这些区域的分子特征及其在治疗结果中的潜在作用。
总之,研究中使用的模型可解释性技术提供了模型预测的可视化表示,使研究人员能够看到模型对肿瘤图像的哪些部分有信心预测治疗反应。虽然这些热图并不能完全解释模型的内部运作,但它们提供了一定程度的透明度,有助于了解模型的决策过程,并确定需要进一步研究的领域。
















 5892
5892











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









