







小罗碎碎念
在癌症研究领域,肿瘤异质性严重阻碍了临床治疗的进展,而单细胞RNA测序(scRNA-seq)技术为深入探究肿瘤细胞的奥秘提供了有力手段。
本文作者利用Microwell-seq技术,精心构建了涵盖303,351个细胞的泛癌单细胞转录组景观,其中包含了中国发病率和死亡率较高的多种肿瘤类型。
通过对恶性细胞、癌前细胞以及肿瘤微环境中各类细胞的深入剖析,清晰地揭示了它们的异质性表达模式。这一成果不仅有助于我们更深刻地理解肿瘤的发生和发展机制,也为后续的药物研发奠定了坚实基础。
针对传统药物筛选方法效率低下的问题,作者创新性地引入了名为Shennong的深度学习框架。该框架巧妙地整合了scRNA-seq数据与扰动数据,能够在单细胞水平上精准预测细胞对药物的反应,高效筛选出潜在的抗癌药物靶点,并全面评估药物对组织的潜在损伤效应。

通过实际应用,Shennong成功筛选出一系列具有潜力的抗癌药物,其中不乏已获FDA批准但正进行新适应症临床试验的药物,以及尚未获批但展现出抗肿瘤活性的候选药物。同时,它对药物组织损伤效应的预测也与实际临床情况高度吻合。
尽管这项研究取得了显著进展,但仍存在一些不可忽视的局限性。目前的研究高度依赖特定的扰动数据,这些数据在收集平台和样本类型上存在一定的局限性,可能导致结果的偏差,影响其广泛应用。此外,在预测药物副作用方面,该框架也存在不足,无法全面涵盖所有潜在的不良反应。
未来的研究需要进一步拓展和优化扰动数据集,纳入更多类型的数据,如配体和siRNAs等,同时加强实验验证,以提升预测的准确性和可靠性,推动医学AI在癌症治疗领域的深度应用。
项目复现流程
(注意,本篇推送仅概述复现流程,梳理代码架构,具体的复现教程请关注后续推送)
1、配置环境
2、准备数据
3、数据预处理
4、训练模型
5、预测与可视化
部分复现效果图
这组图通过可视化的方式展示了单细胞数据在不同分类标准下的分布情况。
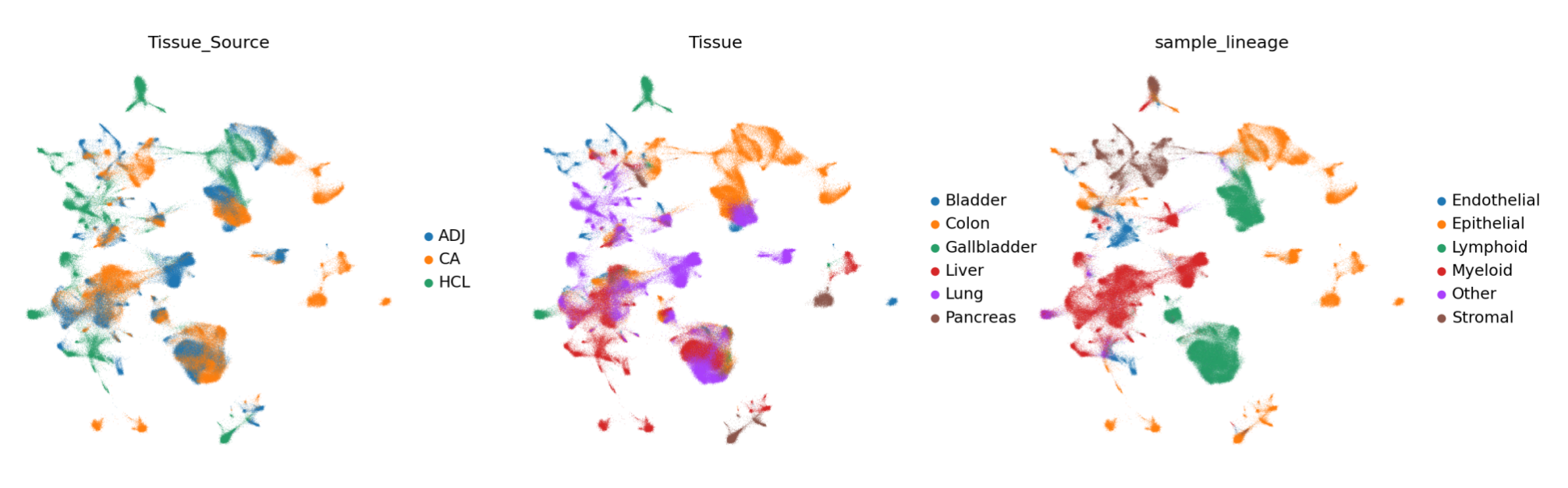
通过下面这张图,可以直观地看出不同细胞类型在二维空间中的分布情况,相近的细胞类型在图中位置较为靠近,有助于研究人员分析细胞类型之间的关系和差异,比如哪些细胞类型在分布上具有相关性,哪些细胞类型则相对独立 。
知识星球
如需获取推文中提及的各种资料,欢迎加入我的知识星球!
星球用户可以直接获取本篇推送的pdf版本,并且可以在星球中向我提问!
一、文献速览
本文通过构建泛癌单细胞转录组图谱,深入分析肿瘤细胞和肿瘤微环境细胞的异质性表达模式,并开发了深度学习框架Shennong进行单细胞水平的抗癌药物筛选,为癌症研究和药物开发提供了新的思路和方法。
- 研究背景:肿瘤异质性对癌症进展和治疗耐药性有重要影响。单细胞RNA测序(scRNA-seq)可揭示肿瘤内异质性,人工智能在癌症研究和药物创新中发挥重要作用,但现有药物反应预测工具存在局限性。
- 构建泛癌单细胞图谱:使用Microwell-seq技术对7种常见实体癌的肿瘤及相邻组织进行单细胞测序,构建包含303,351个细胞的泛癌单细胞图谱。通过无监督聚类识别出51个主要细胞簇,分属5个主要细胞谱系,发现细胞类型在肿瘤类型和个体患者间存在差异。
- 分析细胞异质性表达模式
- 恶性和癌前细胞:整合正常组织scRNA-seq数据,通过多种方法准确识别恶性和癌前细胞。发现癌前细胞可能是癌变早期阶段,具有与肿瘤细胞相似的代谢和细胞间通讯特征,且在基因调控模式上表现出介于正常和恶性细胞之间的中间状态。
- 癌症相关成纤维细胞和内皮细胞:对基质细胞和内皮细胞进行重新聚类分析,发现肿瘤和相邻组织中的基质细胞CNV得分无显著差异,但高于正常基质细胞;癌症相关内皮细胞存在氧化磷酸化下调和细胞增殖相关基因集富集的现象。
- 开发Shennong框架:提出Shennong深度学习框架,整合scRNA-seq数据和扰动数据,在单细胞水平预测细胞对药物的反应、筛选潜在抗癌药物靶点和评估药物潜在组织损伤效应。该框架在训练和预测中表现出良好的性能,通过10折交叉验证证明其稳健性。
- 药物筛选与分析
- 识别抗癌药物和潜在靶点:应用Shennong框架筛选出多种具有抗癌潜力的药物,如azacitidine、irinotecan等已获批药物和parbendazole等候选药物,确定了它们在不同癌细胞类型中的潜在靶点和作用机制,并通过细胞实验验证了部分药物的抗癌活性。
- 评估组织损伤效应:Shennong框架预测了一些药物的组织损伤效应,如vemurafenib对正常胰腺上皮细胞、lopinavir和GSK-690693对正常肝脏细胞的潜在损伤,这些预测与临床报道相符。
- 研究讨论:本研究利用Microwell-seq构建泛癌单细胞图谱,为癌症研究提供了资源。Shennong框架在药物筛选和评估方面展现出潜力,但研究存在局限性,如依赖扰动数据、对药物副作用预测不全面等。未来需更大更丰富的数据集和实验验证来改进。
二、构建泛癌单细胞图谱
2-1:单细胞RNA测序实验和生物信息学工作流程
实验流程主要包括样本采集、单细胞测序、构建泛癌单细胞图谱以及后续分析等步骤。
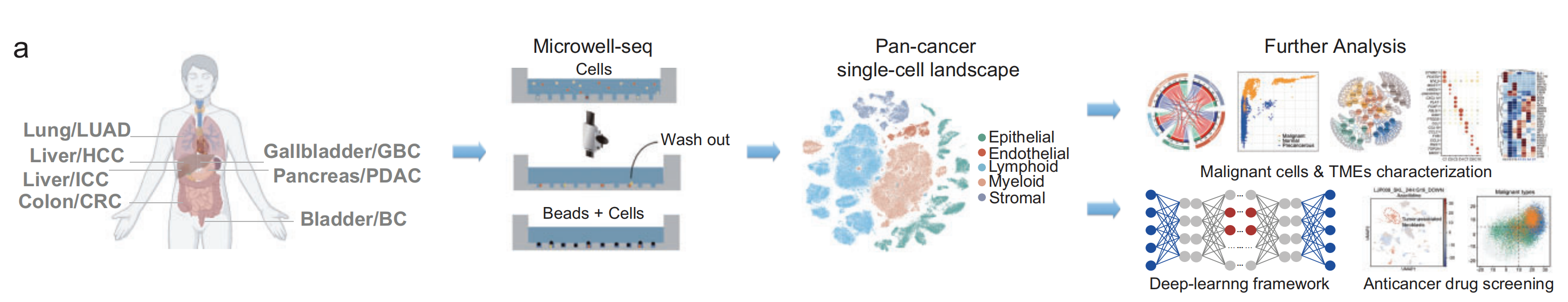
- 样本采集:从人体的多个部位采集肿瘤样本,包括肺(LUAD,肺腺癌)、肝脏(HCC,肝细胞癌;ICC,肝内胆管癌)、结肠(CRC,结直肠癌)、胆囊(GBC,胆囊癌)、胰腺(PDAC,胰腺导管腺癌)、膀胱(BC,膀胱癌)。
- 单细胞测序:使用Microwell-seq技术,先将细胞放入微孔中,冲洗掉多余细胞后,让微珠与细胞结合,进行单细胞RNA测序。
- 构建泛癌单细胞图谱 :对测序后的数据进行整合分析,构建出泛癌单细胞图谱,图谱中标注了不同类型的细胞,如上皮细胞、内皮细胞、淋巴细胞、髓细胞和基质细胞。
- 后续分析:一方面对恶性细胞和肿瘤微环境(TMEs)进行特征分析;另一方面利用深度学习框架进行抗癌药物筛选。
2-2:患者的肿瘤类型与分析细胞数量
(b) 堆积条形图展示了每种肿瘤类型和每位患者的分析细胞数量,饼图展示了肿瘤(CA)和相邻(ADJ)组织中分析细胞的百分比。
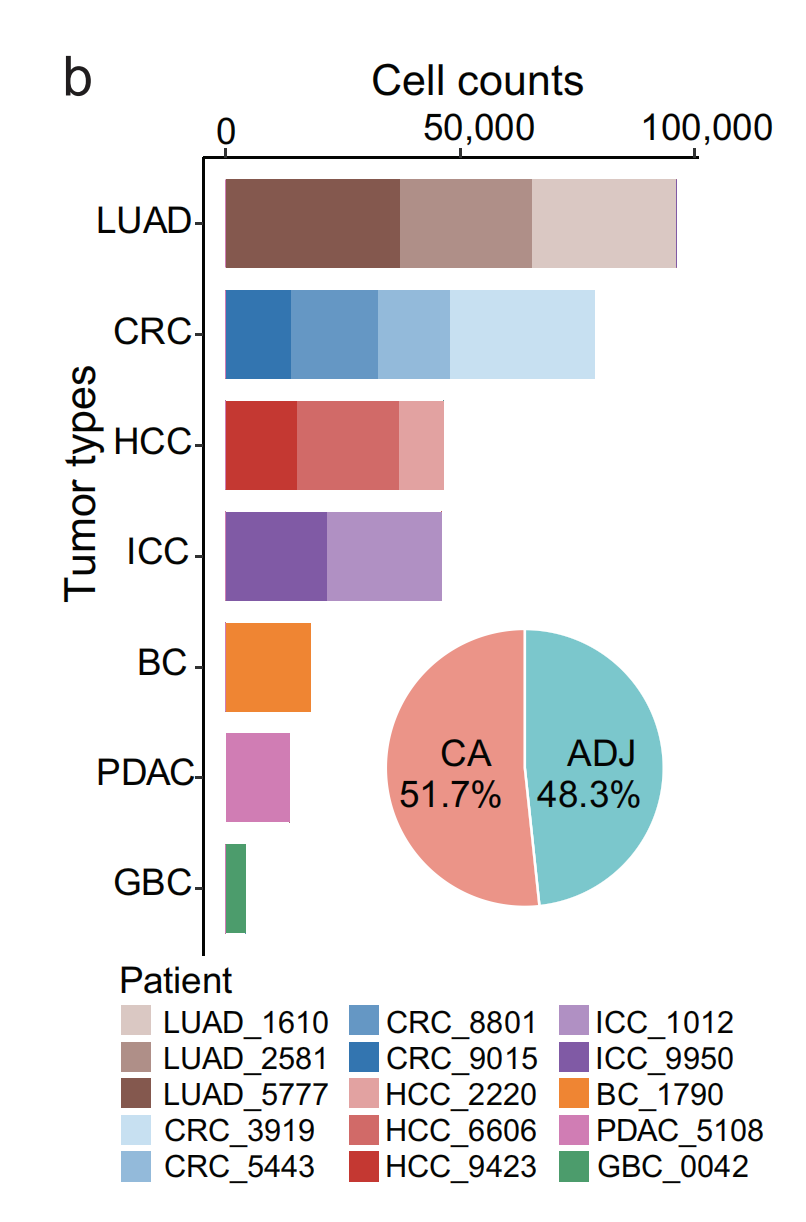
- 堆积条形图:展示了不同肿瘤类型(LUAD肺腺癌、CRC结直肠癌、HCC肝细胞癌、ICC肝内胆管癌、BC膀胱癌、PDAC胰腺导管腺癌、GBC胆囊癌 )中分析的细胞数量,每种肿瘤类型的细胞又按不同患者区分。其中,LUAD分析的细胞总数最多,GBC分析的细胞总数最少。
- 饼图:展示了肿瘤组织(CA,占51.7%)和相邻组织(ADJ,占48.3%)中分析细胞的比例 。
2-3:t-SNE可视化
© 对泛癌单细胞图谱中的303,351个单细胞进行t-SNE可视化,根据细胞簇身份(n = 51)和肿瘤类型(n = 7)进行着色。
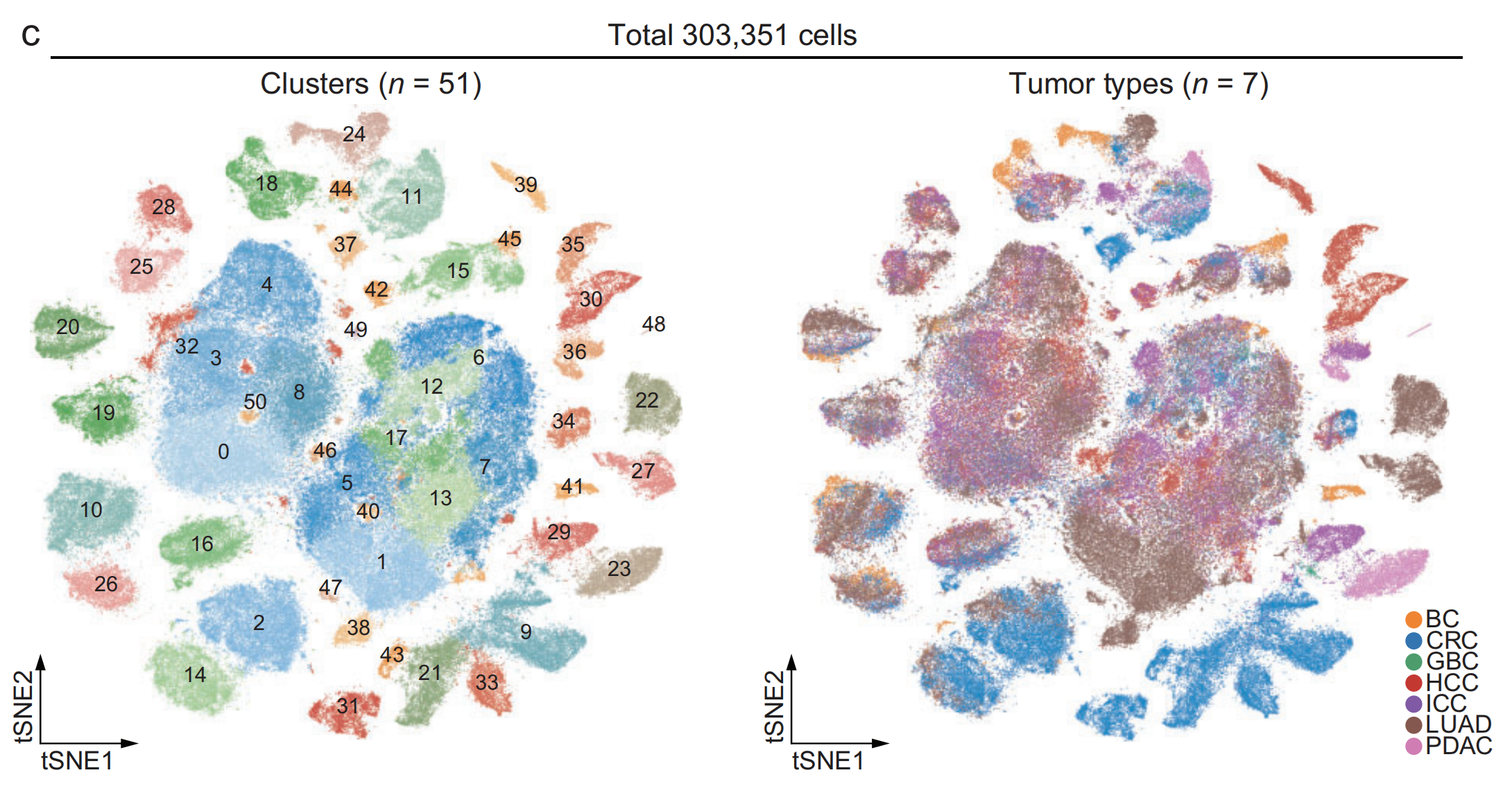
- 左侧图:依据细胞簇身份对303,351个细胞进行标记,共51个细胞簇,每个细胞簇用不同颜色区分,并标有数字编号。这能直观呈现不同细胞簇在图谱中的分布情况,可用于分析细胞簇之间的关系和特征。
- 右侧图:按照7种肿瘤类型(BC膀胱癌、CRC结直肠癌、GBC胆囊癌、HCC肝细胞癌、ICC肝内胆管癌、LUAD肺腺癌、PDAC胰腺导管腺癌 )对细胞进行着色,每种肿瘤类型对应一种颜色。通过该图可看出不同肿瘤类型的细胞在图谱中的分布区域,有助于对比不同肿瘤细胞的分布差异。
2-4:细胞分布
(d) 层次聚类树展示了51个细胞簇之间的相似性,直方图展示了每个细胞簇的组织来源百分比。
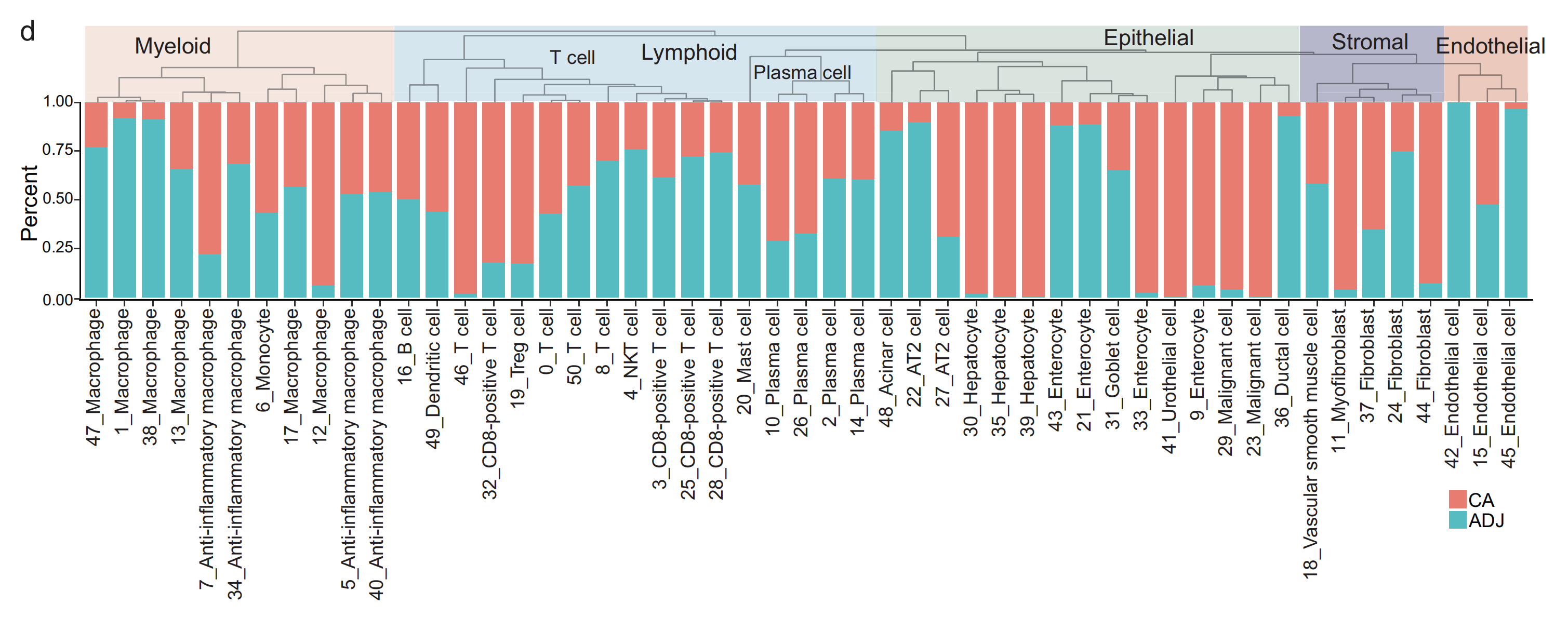 这张图展示了不同细胞类型在肿瘤组织(CA)和相邻组织(ADJ)中的比例分布情况:
这张图展示了不同细胞类型在肿瘤组织(CA)和相邻组织(ADJ)中的比例分布情况:
- 细胞类型分类:图上方将细胞类型分为髓系(Myeloid)、淋巴细胞系(Lymphoid,包括T细胞和浆细胞等)、上皮细胞(Epithelial)、基质细胞(Stromal)和内皮细胞(Endothelial)。
- 具体细胞亚类及比例:每一列代表一种具体的细胞亚类,如各种巨噬细胞(Macrophage)、T细胞亚型(如CD8 - positive T cell等)、浆细胞(Plasma cell)等。红色部分表示肿瘤组织(CA)中该细胞亚类所占比例,蓝色部分表示相邻组织(ADJ)中该细胞亚类所占比例。通过对比不同细胞亚类在CA和ADJ中的比例,可以分析肿瘤微环境中细胞组成的差异 。
三、通过单样本分析鉴定恶性细胞和癌前细胞
3-1:不同维度的数据分析癌症相关细胞
(a) 堆积条形图展示了泛癌单细胞图谱和人类细胞图谱(HCL)中每种组织类型和来源的样本数量,饼图展示了肿瘤(CA)、相邻(ADJ)和正常(HCL)组织中分析样本的百分比。
(b) 不同组织来源的肿瘤微环境(TMEs)中5种主要细胞谱系之间的细胞相互作用。
© 肿瘤微环境中5种主要细胞谱系之间的相互作用。弧线的长度代表预测的相互作用次数。
(d) 对来自患者ICC_1012的24,628个单细胞进行UMAP可视化(左上角),分别根据CD151和EPCAM富集情况进行着色(右上角)、组织来源(左下角)和细胞谱系(右下角)。
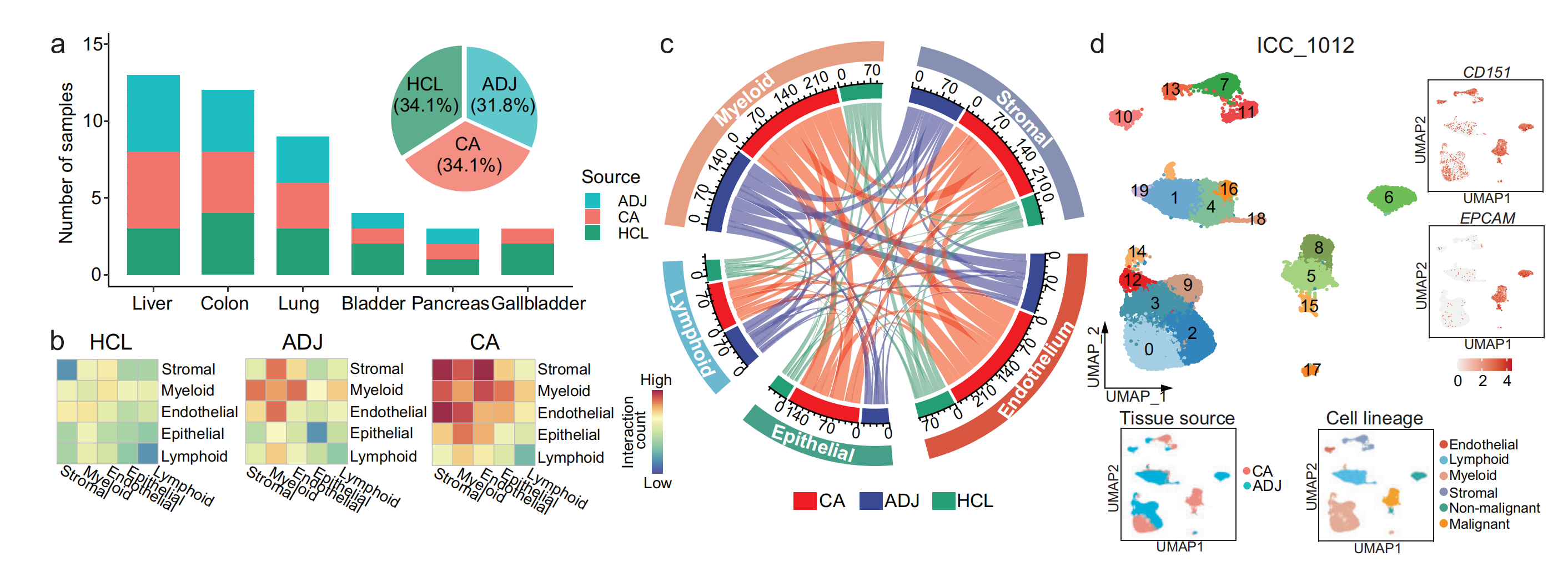
- 图a:左侧堆积条形图显示了肝脏、结肠、肺、膀胱、胰腺、胆囊这些部位中,来自肿瘤组织(CA)、相邻组织(ADJ)和人类细胞图谱(HCL)的样本数量分布。右侧饼图表明在所有分析样本中,CA、ADJ、HCL分别占比34.1%、31.8%、34.1%。
- 图b:展示了HCL、ADJ、CA中不同细胞谱系(基质细胞、髓细胞、内皮细胞、上皮细胞、淋巴细胞)之间相互作用的热图。颜色深浅代表相互作用计数的高低,可直观看出不同组织来源中细胞谱系间相互作用的差异。
- 图c:以弦图展示了不同组织来源(CA、ADJ、HCL)中5种主要细胞谱系(髓细胞、淋巴细胞、上皮细胞、基质细胞、内皮细胞)之间的相互作用。线条的粗细和颜色表示相互作用计数的多少,能清晰呈现不同细胞谱系在不同组织来源间的复杂联系。
- 图d:对患者ICC_1012的24628个单细胞进行UMAP可视化。左图根据细胞簇展示分布;右上角两个小图分别根据CD151和EPCAM富集情况着色;左下角小图按组织来源(CA、ADJ)区分;右下角小图按细胞谱系(内皮细胞、淋巴细胞、髓细胞、基质细胞、非恶性细胞、恶性细胞)标记,有助于分析该患者细胞的特征和分布。
3-2:多方面分析
这一部分围绕患者ICC_1012及泛癌数据,从拷贝数变异(CNV)、细胞相互作用、代谢途径等方面进行了分析。
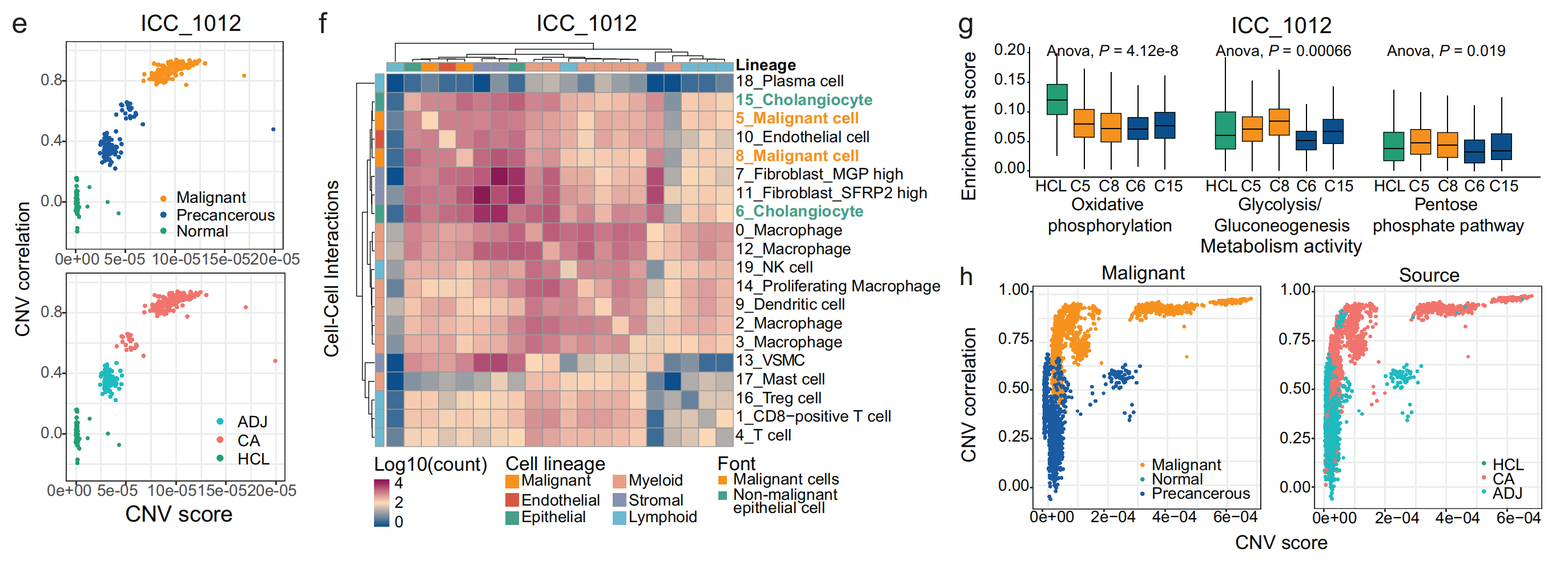
- 图e:针对患者ICC_1012,展示上皮细胞的CNV分数(x轴)和CNV相关性(y轴)。上图按细胞恶性类型分类,橙色为恶性细胞,蓝色为癌前细胞,绿色为正常细胞;下图按组织来源区分,红色为肿瘤组织(CA),蓝色为相邻组织(ADJ),绿色为人类细胞图谱(HCL)中的组织 ,可用于观察不同类型和来源细胞的CNV特征。
- 图f:是患者ICC_1012细胞簇之间相互作用的热图。左侧和顶部标注了不同细胞谱系,颜色深浅代表细胞间相互作用计数的对数(Log10(count)),能直观呈现不同细胞簇之间相互作用的强度和模式。
- 图g:通过箱线图对比了人类细胞图谱(HCL)中的正常组织和患者ICC_1012上皮细胞簇在“氧化磷酸化”、“糖酵解/糖异生”和“磷酸戊糖途径”这三条代谢途径的富集分数。图上还标注了方差分析(Anova)的P值,说明不同组之间在这些代谢途径上存在显著差异。
- 图h:左图为泛癌数据中所有上皮细胞的CNV分数和CNV相关性,并按细胞恶性类型分类;右图同样展示CNV分数和CNV相关性,但按组织来源区分。通过这两张图可对泛癌上皮细胞的CNV情况进行整体分析。
四、材料与方法
4-1:患者与样本采集
伦理与合规性
- 样本来源:手术切除的肿瘤组织(CA)及癌旁组织(ADJ),确保样本代表性与临床相关性。
样本处理流程
- 样本保存:术后立即置于4°C DMEM培养基中,2小时内完成单细胞测序,最大限度保持细胞活性。
- 临床信息:患者详细临床数据见表S1,涵盖肿瘤类型(如肺癌、肝癌等)及患者基本信息。
4-2:单细胞测序与数据处理
使用Microwell-seq技术进行高通量单细胞RNA测序,具体流程包括:
- 细胞裂解与逆转录
- 双链cDNA合成与扩增
- 转座酶片段化与文库构建
- 定制测序引物(表S4)优化数据质量
- MGI DNBSEQ-T7平台测序
数据分析流程
- 数据预处理:比对至人类基因组GRCh38,过滤低质量细胞(如线粒体基因占比过高或低UMI计数的细胞)。
- 降维与聚类:
- 使用Seurat和Scanpy工具进行PCA、t-SNE/UMAP降维。
- 基于基因表达谱将细胞划分为5大类(上皮细胞、基质细胞、内皮细胞、淋巴系细胞、髓系细胞)。
- 差异基因分析:识别细胞类型特异性标志基因(如EPCAM标记上皮细胞,CD3D标记T细胞)。
4-3:恶性细胞鉴定方法
多策略联合鉴定
- 表达谱分析:
- 聚类后筛选上皮细胞,对比肿瘤与正常组织的差异基因(如癌基因高表达)。
- 拷贝数变异(CNV)推断:
- 使用inferCNV包,以正常组织上皮细胞为参考,计算肿瘤细胞的CNV评分(恶性细胞CNV显著高于正常)。
- 降维验证:
- 恶性细胞在降维空间(如UMAP)中形成独立簇,与正常细胞分离(图2d-e)。
意义
- 多方法互补提高恶性细胞鉴定的准确性,避免单方法偏差(如仅依赖表达谱可能误判炎性细胞)。
- 支持后续药物筛选的靶向性(如区分恶性细胞与癌前细胞)。
4-4:数据整合与验证
-
跨数据集验证:
- 整合公共数据集(如Tabula Sapiens、第三方单细胞数据),验证泛癌图谱的泛化能力。
- 使用第三方数据(如LUAD数据集)验证Shennong框架的稳健性(图5k-l)。
-
质量控制:
- 细胞批次效应校正:通过Harmony或CCA算法整合多患者数据,确保聚类不受批次干扰。
- 代谢通路富集分析:验证恶性细胞的代谢重编程特征(如糖酵解上调,氧化磷酸化抑制)。
4-5:技术亮点与挑战
创新点
- Microwell-seq的高通量优势:单次实验分析30万+细胞,捕捉罕见细胞亚群(如癌前细胞)。
- 多组学整合:CNV、代谢通路、细胞通讯网络联合分析,全面解析肿瘤微环境。
- Shennong框架的通用性:支持跨癌种药物敏感性预测,适用于老药新用与新药开发。
挑战
- 数据规模大:需高性能计算资源处理PB级数据。
- 临床转化瓶颈:体外实验与临床疗效的差异(如GSK-126在临床试验中因剂量限制疗效不足)。
4-6:总结
材料与方法部分体现了研究的严谨性与技术创新:
- 样本处理与测序流程的标准化确保数据可靠性;
- 多策略恶性细胞鉴定提升结果的可信度;
- 高通量数据与深度学习框架的结合,为精准药物筛选提供技术基础。
这些方法不仅支持了“Shennong”框架的开发,也为后续肿瘤异质性研究和临床转化奠定了数据基础。
五、Shennong分析流程解读
5-1:框架概述
Shennong是一个基于深度学习的单细胞水平药物扰动预测框架,旨在通过整合单细胞转录组数据和大规模药物扰动数据库,实现抗癌药物的计算机模拟筛选。
其核心流程分为三个阶段(图4a),具体如下:
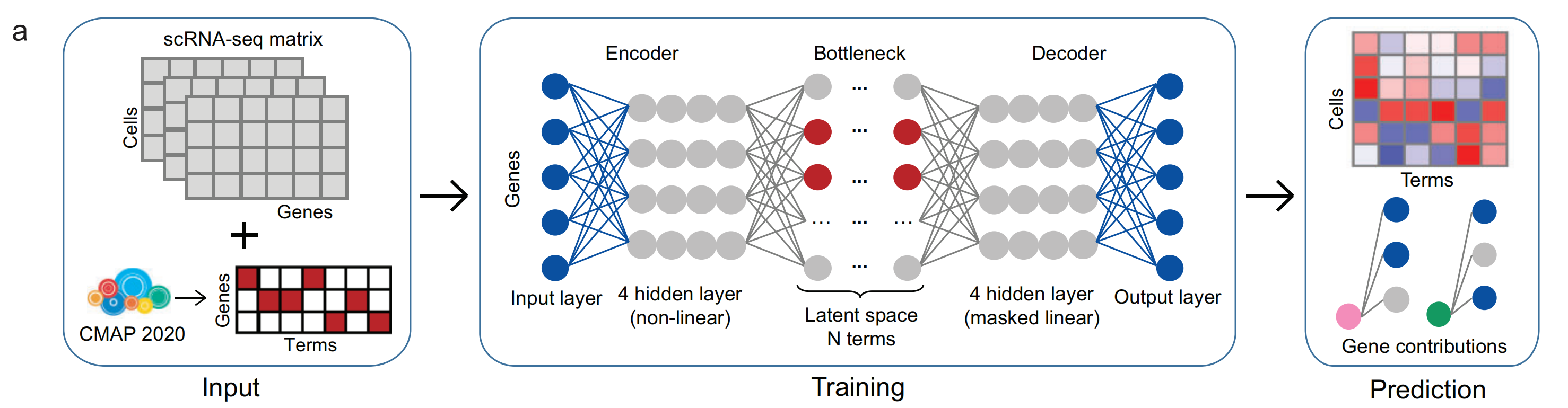
- 输入(Input):将scRNA-seq矩阵与CMAP 2020(连接图谱2020)数据相结合。scRNA-seq矩阵记录了细胞(Cells)和基因(Genes)的相关信息,CMAP 2020数据则转换为基因与术语(Terms)相关的矩阵。
- 训练(Training):通过一个包含编码器(Encoder)、瓶颈层(Bottleneck)和解码器(Decoder)的深度学习模型进行训练。编码器有4个非线性隐藏层,用于提取输入数据特征;瓶颈层是潜在空间(Latent space),包含N个术语;解码器有4个掩码线性隐藏层,用于将潜在空间的特征转换回可解释的输出。
- 预测(Prediction):模型训练完成后,对细胞和术语进行分析,得到基因贡献(Gene contributions)结果,以热图和连线图的形式展示,可用于探索基因在不同细胞中的作用和贡献 。
5-2:分析流程详解
阶段一:数据整合与特征提取
- 输入数据:
- 单细胞RNA测序数据:来自泛癌图谱的388,646个细胞(涵盖肿瘤、癌旁及正常组织)。
- 扰动数据:预处理后的基因集文件(
.gmt格式),每个条目(term)对应特定化合物和实验条件下的差异表达基因集合(源自CMap数据库)。
- 关键操作:
- 将单细胞表达矩阵与扰动数据结合,生成扰动变化特征矩阵,每个term代表一种药物作用的基因表达模式。
阶段二:构建细胞扰动预测模型
- 模型架构:
- 变分自编码器(CVAE):基于公开模型expiMap改进,包含4层编码器和4层解码器。
- 编码器:非线性结构,灵活捕捉单细胞数据的异质性。
- 解码器:线性结构(带掩码),确保模型可解释性。
- 潜在空间维度:与term数量一致,每个维度对应一个term的潜在特征。
- 变分自编码器(CVAE):基于公开模型expiMap改进,包含4层编码器和4层解码器。
- 训练策略:
- 超参数优化:通过预训练确定最佳参数(如
alpha_kl=0.005,alpha=0.95, 隐藏层维度为512)。 - 损失函数:结合KL散度正则化和稀疏性约束,平衡模型复杂性与泛化能力。
- 超参数优化:通过预训练确定最佳参数(如
阶段三:药物影响评估与机制解析
- 影响评分计算:
- 基于训练好的模型,计算每个term对单细胞的影响评分(Influence Term Score),反映药物扰动对细胞的潜在效应。
- Bayes因子分析:筛选显著差异的term(|log-Bayes因子|>2.3),用于区分细胞类型特异性药物响应。
- 基因贡献解析:
- 从解码器权重中提取每个term内基因的贡献度,排序后识别关键靶基因(如volasertib通过抑制PLK1和BCL2L1诱导凋亡)。
5-3:泛癌图谱中的应用
- 数据划分:
- 训练集:346,129个细胞(90%数据),覆盖6种组织类型。
- 预测集:42,517个细胞(10%数据),用于验证模型泛化能力。
- 结果验证:
- 潜在空间可视化:预测集细胞在潜在空间中与训练集高度整合,聚类结果与细胞类型注释一致(图4b)。
- 跨数据集稳健性:在第三方数据集(如LUAD、HCC)中,30%-45%的显著差异term与原始预测重叠(图5k)。
5-4:技术亮点与资源开放
- 创新点:
- 高通量整合:同时分析数十万单细胞,捕捉罕见细胞亚群(如癌前细胞)的响应。
- 可解释性设计:线性解码器与基因贡献排序,揭示药物作用的分子机制。
- 跨癌种预测:支持泛癌药物敏感性评估及组织毒性预测(如venurafenib的胰腺毒性)。
- 数据与代码:
- 数据公开:原始测序数据存于GSA-Human(HRA006591),处理后的矩阵及注释发布于Figshare(https://figshare.com/s/ac34f719115943d1d46c)。
- 代码开源:分析流程与模型代码共享于GitHub(https://github.com/PeijingZhang/Shennong)。
5-5:应用与挑战
- 临床潜力:
- 老药新用:预测抗寄生虫药parbendazole、抗HIV药物lopinavir的抗癌活性。
- 毒性预警:识别GSK-690693的肝损伤风险,与临床试验终止结果一致。
- 局限性:
- 依赖批量扰动数据:可能忽略细胞类型特异性响应。
- 系统性副作用预测不足:需结合代谢组学或蛋白组学数据优化。
六、Shennong深度学习框架
6-1:框架概述
Shennong 是一个基于深度学习的单细胞水平抗癌药物筛选框架,通过整合泛癌单细胞转录组数据和大规模药物扰动数据库(如CMap),实现以下功能:
- 预测药物对特定细胞簇的敏感性(如恶性细胞、癌相关成纤维细胞)。
- 评估药物对正常组织的潜在毒性(如肝细胞、胰腺细胞损伤)。
- 解析药物作用机制(通过基因贡献度排序识别关键靶点)。
核心优势
- 高分辨率:单细胞级别预测,捕捉罕见细胞亚群响应。
- 可解释性:线性解码器设计支持基因贡献度分析。
- 泛癌适用性:支持多癌种数据整合与跨数据集验证。
6-2:环境配置
系统要求
- Python ≥ 3.8
- GPU支持(推荐NVIDIA CUDA 11.6+)
依赖安装
# 创建虚拟环境(可选)
conda create -n shennong python=3.8
conda activate shennong
# 安装核心依赖
pip install scanpy==1.9.2 scarches==0.5.7 torch==1.13.1 pandas==1.5.3 numpy==1.23.5 gdown==4.6.3
6-3:数据准备
输入数据
-
单细胞RNA数据(
scRNA.h5ad):需包含标准化后的表达矩阵和细胞注释(如细胞类型、组织来源)。- 示例数据:可从FigShare下载预处理好的泛癌数据集。
-
扰动数据(
.gmt文件):包含药物或化合物对应的差异基因集。-
默认数据:下载预处理的CMap扰动数据:
wget http://bis.zju.edu.cn/shennong/data/perturbation.gmt wget http://bis.zju.edu.cn/shennong/data/perturbation.gmt.h5ad
-
数据目录结构
Shennong/
├── data/
│ ├── perturbation.gmt # 扰动基因集文件
│ └── perturbation.gmt.h5ad # 预处理后的扰动数据
├── script/
│ ├── preprocess.py # 预处理脚本
│ ├── train.py # 训练脚本
│ └── predict.py # 预测脚本
└── example/
└── example.ipynb # Jupyter示例
6-4:完整流程
数据预处理
目的:整合单细胞数据与扰动数据,生成模型输入格式。
python script/preprocess.py \
adata_train.h5ad \ # 输入单细胞数据
data/perturbation.gmt \ # 扰动基因集
data/perturbation.gmt.h5ad \ # 预处理的扰动数据
adata_train_gmt.h5ad # 输出文件
输出:adata_train_gmt.h5ad,包含整合后的特征矩阵。
模型训练
目的:构建条件变分自编码器(CVAE),学习药物扰动模式。
python script/train.py \
adata_train_gmt.h5ad \ # 预处理后的数据
model_train/ \ # 模型保存路径
Tissue_Source # 批次校正键(如组织来源)
关键参数:
alpha_kl=0.005:KL散度权重。alpha=0.95:稀疏性约束权重。hidden_layer_sizes=512:隐藏层维度。
预测与可视化
目的:预测新数据集中的药物响应,并可视化潜在空间。
# 预测新数据
python script/predict.py \
adata_train_gmt.h5ad \ # 训练数据
model_train/ \ # 训练模型路径
adata_predict.h5ad \ # 待预测数据
adata_predict_gmt.h5ad \ # 预测输出
model_predict/ # 预测模型保存路径
# 可视化(通过Jupyter)
jupyter lab example/example.ipynb
可视化内容:
- 潜在空间UMAP图(细胞按药物响应评分着色)。
- 基因贡献度点图(显示每个term的关键靶基因)。
6-5:结果解读
-
药物敏感性预测:
- 高评分term表示该药物对目标细胞簇有显著作用。
- 示例:
volasertib在肝癌细胞中评分高,提示其通过抑制PLK1诱导凋亡。
-
组织毒性评估:
- 正常细胞簇中高评分term提示潜在毒性。
- 示例:
venurafenib在胰腺细胞中评分高,与临床肝毒性报道一致。
-
机制解析:
- 解码器权重排序前10的基因视为关键靶点。
- 示例:
azacitidine的贡献基因包括DNMT1和CDKN2A,验证其去甲基化机制。
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!


















 1023
1023

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









