






一.SAGDTI: self-attention and graph neural network with multiple information representations for the prediction of drug-target interactions
具有多种信息表示的自我注意和图形神经网络,用于预测药物-靶点相互作用,2023.8 二区
SAGDTI是第一种在SMILES序列和三维结构图的输入形式中充分考虑药物和靶标的独特分子属性表示的方法。现有的深度学习方法总是简单地构建通过连接特征表示来针对相关目标进行药物不能充分表达相互作用。为了准确捕获化合物和受体的独特分子信息,设计了分子Transformer模块来改善药物化合物中原子之间的相对元素信息和靶蛋白结合口袋的三维结构特征。由于其自注意性,所提出的方法是注意力衍生的,由于其自我注意,在聚合分子和生物特征信息的属性表征时提供了很高的可解释性。作者认为AttentionSiteDTI方法等这些模型都捕获了DTI特征,但没有充分利用独特的分子特征属性。
1.模型
SAGDTI 接受三个来自多个分子和生物数据源的输入表示,包括具有相对原子距离的 SMILES 字符串,包含空间信息的蛋白质的图嵌入结合位点以及基于图形的相互作用信息异构网络。
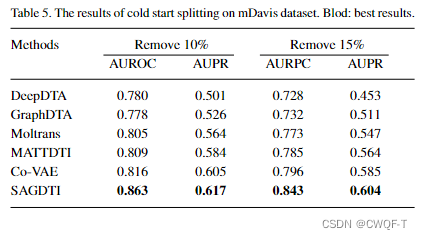
1.嵌入:药物SMILES应用Zeng et al., 2021提出的覆盖原子之间的远距离相对位置信息的方法被嵌入为序列向量(包括令牌嵌入和位置嵌入),为了模拟原子之间的相对距离,使用m种原子之间的相对相关性来描述独特的分子特征,并且设置SMILES字符串中特殊字符(如@,=)之间的距离为0,将SMILES序列的最大长度限制为一个超参数;受蛋白质的图输入表示(Yazdani et al., 2022)的启发,具有与化合物相互作用的结合位点的蛋白质口袋被嵌入到3dgraph中。绘制了单独的图来表示每个结合位点的每个原子a(节点)和原子之间的关系(边)。对于每个原子都创建的大小为k 的特征向量。最后得到每个结合位点的图。故 蛋白质的特征被表示为图,药物的特征被表示为序列。
2.使用基于多头注意力机制(MSA)的transformer模块来提取蛋白图和药物序列的特征,每个药物-靶标对的特征被转换为交互配对图,并且送到2D-CNN中进行原子级的分子属性的表示工作。对于药物的嵌入,将其输入到MSA层中以获取药物中每个原子的注意力分数。每个分子Transformer编码器块使用残差运算进行叠加,增强分子特征提取。为了在原子水平上进一步学习药物-蛋白对的分子属性,将获得的药物及其蛋白质结合位点的特征表示通过进行点积操作来转换为相互作用配对图。再采用CNN层提取交互图中原子的邻接原子的信息。得到药物和蛋白质的分子属性向量。
3.构建图注意力网络GAT,通过图化注意力,以异构网络(矩阵)的形式捕获覆盖多尺度拓扑结构和多样化连接的交互信息:对于每个节点hi,采用掩码关注来计算关注系数,并且只关注节点hi及其一阶相邻节点,还给与它相连的边赋权重。再采用MSA策略,通过采用非线性激活函数对注意力系数进行归一化。通过连续的GAT层提取药物与靶标相互作用信息的复杂特征表示后,利用全局最大池化方法输出 药物和蛋白质的 生物属性向量。
4.最后,使用卷积池(C-P)网络(由交替的卷积层和池化层组成,代码中显示一共三层卷积层,两个归一化层,一个线性层)来给两个获得的属性分配权重,并使用全连接神经网络(FNN)来预测药物-靶点对的交互得分。大多数现有方法只是简单地将学习到的特征表示连接起来或将它们包含在几个FNN层中。这些方法不能完全学习药物与其结合靶标之间的真实相互作用知识。受Zeng et al., 2021对DTI特征建模的启发,应用C-P和FNN层将提取的特征映射到最终的分类输出。首先,将分子属性表示(sxmol)和生物属性表示(XBio)进行左右拼接,得到最终的特征表示。将它输入C-P和FNN层,学习整个DTI特征,并使用Sigmoid函数输出最终的交互评分。
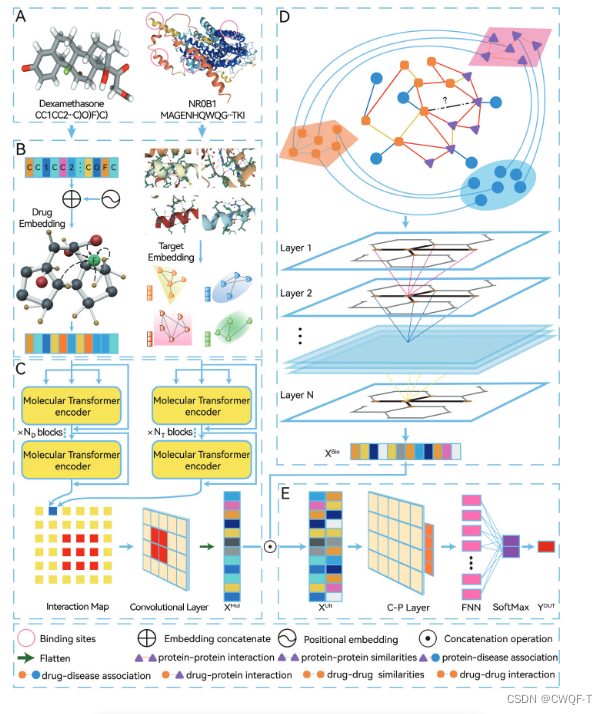
1.药物表示
将SMILES转化为序列向量
2.蛋白质表示
将蛋白质的结合位点提取出来,转化为一个个的3D图
2.实验
应用三种实验设置:a new-target setting, a new-drug setting, and a pairwise setting.对于第一种,蛋白质靶标被随机分成5个相同的子集,其中4个被用于训练,最后一个被用于测试。在训练过程中,SAGDTI模型学习训练数据,其中包含训练靶标和与它相关的所有药物。随后,使用训练好的模型来建议测试蛋白质靶标和所有药物之间的成对相互作用。其他两种设置类似。
此外,五重交叉验证:每个子集都会当成测试集一次。也就是说,测试集:训练集是1:5。
1.数据集
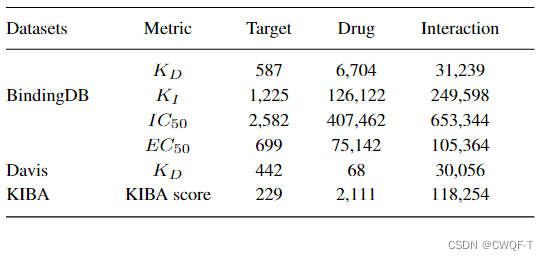
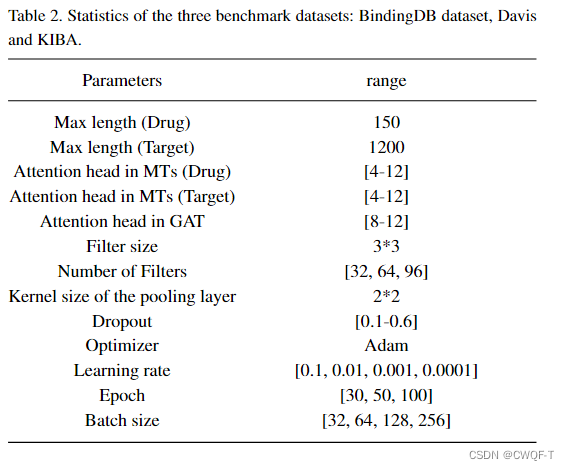
3.结果
1.三种实验设置下的结果
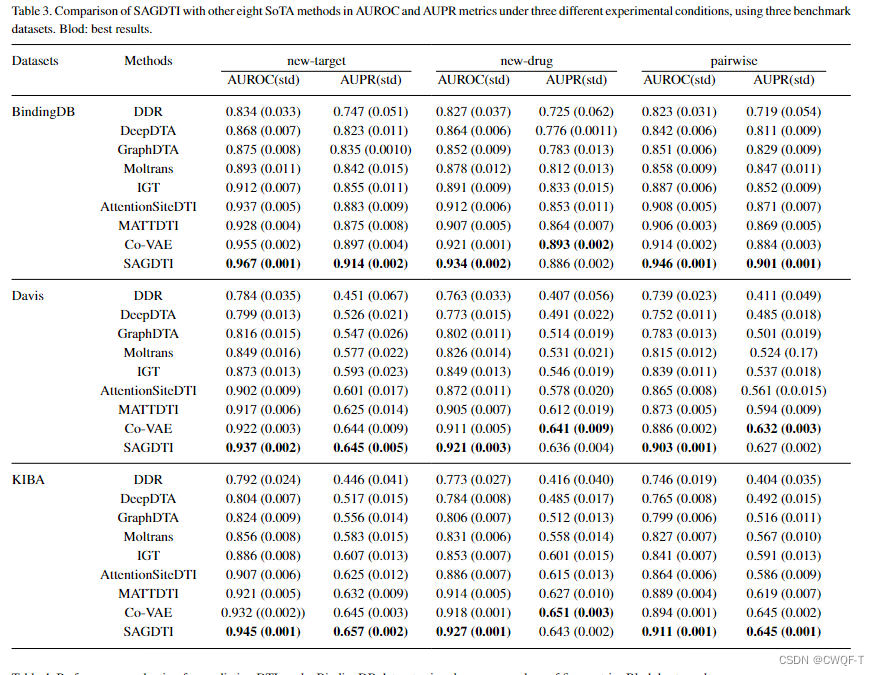
2.五重交叉验证的结果
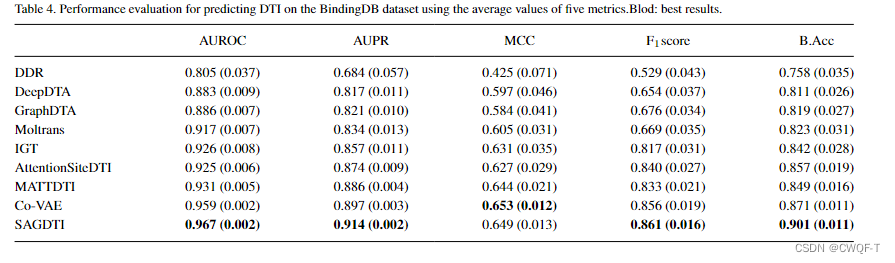
3.使用论文How to approach machine learning-based prediction of drug/compound–target interactions提出的分割数据集的方法,然后随机选择10%或15%的DTI对作为测试样本,并从训练样本中去除所有与之相关的药物和蛋白质。
二.DeepMGT-DTI: Transformer network incorporating multilayer graph information for Drug–Target interaction prediction
结合多层图信息进行药物-靶标相互作用预测的变压器网络 2022.3
1.模型
MCGCN处理了原子序数不一致的药物分子的结构图。结合多层图信息的变压器网络提取了药物分子中的原始信息。分子结构图用于表示药物,以保持原子之间的相互作用。改进的变压器网络融合了图卷积神经网络中层间的特征信息,提取了分子结构的交互作用数据。
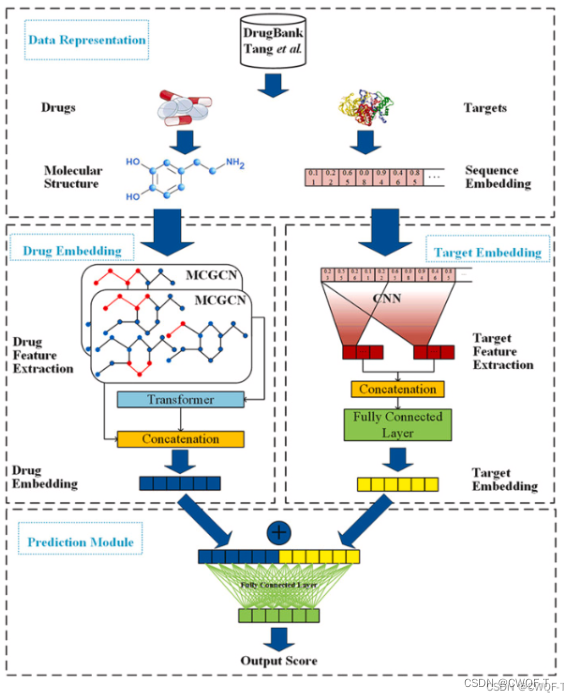
输入药物分子结构信息、靶序列信息和药物-靶点作用关系。药物和目标特征信息分别使用融合图信息的转换器和卷积神经网络提取。最后,我们将药物和靶标特征连接起来,并使用完全连接的神经网络进行DTI预测。
三个模块:数据表示、特征提取和模型预测
1.数据表示:
药物:每种药物特有的分子化学结构表示为图,其顶点和边分别表示药物的原子和化学键。每个药物分子都使用特征矩阵和邻接矩阵表示。特征矩阵的每一行对应于一个原子的属性。
蛋白质:随机初始化一个26*20的查找表,表中的值是氨基酸序列中每个氨基酸出现的概率。基于查找表来构建一个嵌入矩阵,长度是蛋白质序列中的最大长度,设置为2500,宽度是查找表的宽度。???
2.特征提取:
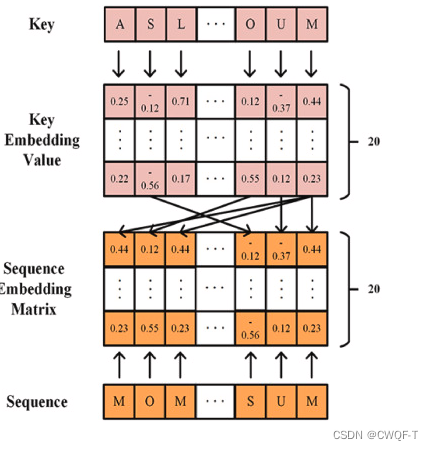
(1)药物:具有融合多层图信息的Transformer由分子互补图卷积神经网络(MCGCN)和改进的Transformer组成。该网络以药物的分子结构为输入。MCGCN提取药物分子图信息,其各层的特征也输入到Transformer中。在Transformer中使用不同的多头注意力机制进行进一步的特征提取。通过进一步提取MCGCN中不同隐藏层的特征,保留了药物分子图中原子间边的信息,丰富了药物分子的特征,克服了GCN忽略节点间关系的缺点。使用MCGCN通过在原始药物分子图中添加互补图来确保每个药物分子的邻接矩阵和特征矩阵具有相同的大小,其中原始图和互补图是独立的。MCGCN有两个隐藏层,每个隐藏层后跟一个ReLU激活函数,并在MCGCN的末尾使用最大池化来降低数据的维度。互补操作后,药物的分子图可以表示为
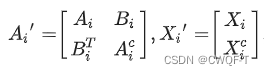
B表示第i种药物的原始图和补充图之间的连接矩阵,以及A、X是邻接矩阵和互补后的特征矩阵.
此外,改进了编码器中的多头注意力机制,MCGCN不同隐藏层的药物特征信息采用两个不同的多头注意力机制进行处理,多头注意力机制处理的特征向量通过concat运算连接并馈送到层归一化部分。每个编码器层还包含一个完全连接的前馈网络和一个两层线性变换。两层线性变换使用 ReLU 激活函数。
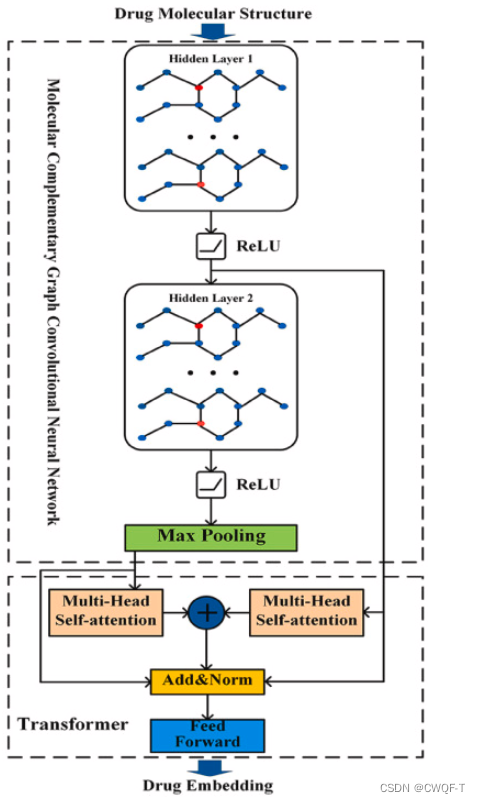
(2)蛋白质:使用CNN捕获从氨基酸序列中提取残基信息,氨基酸序列小的用0填充。三个卷积核大小不同,每个卷积层之后,使用 ELU 激活函数,再使用最大池化。将最大池化后的结果馈送到全连接层,以获得目标序列的嵌入表示。
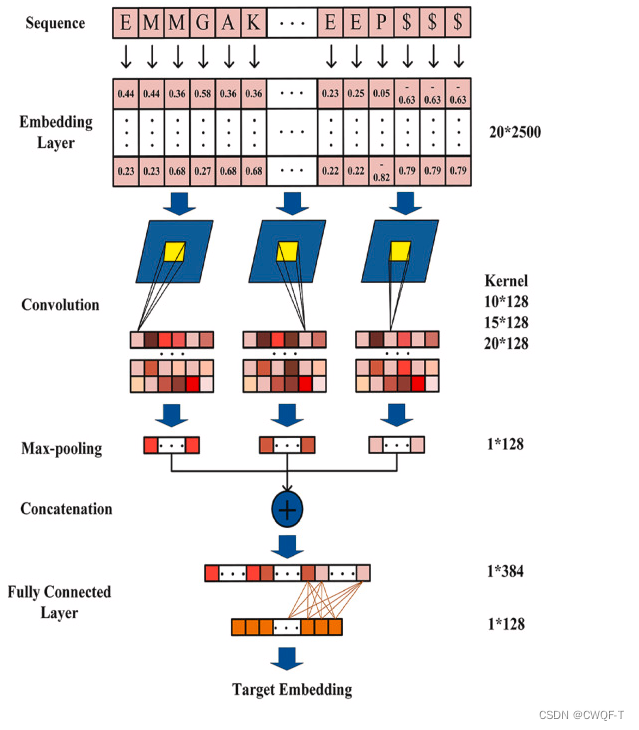
3.预测模块:将药物和蛋白质的嵌入向量串联起来输入全连接层中来预测。
1.药物表示
SMILES经过MCGCN得到图表示
2.蛋白质表示
氨基酸序列
2.实验
1.数据集
药物的靶蛋白序列和SMILES序列分别来自KEGG数据库和PubChem数据库 。用了与PubChem数据库中的Delta变体和阿尔茨海默病相关的DTI数据。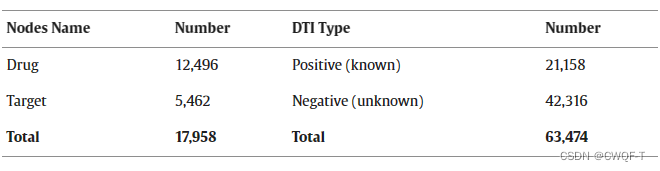
3.结果
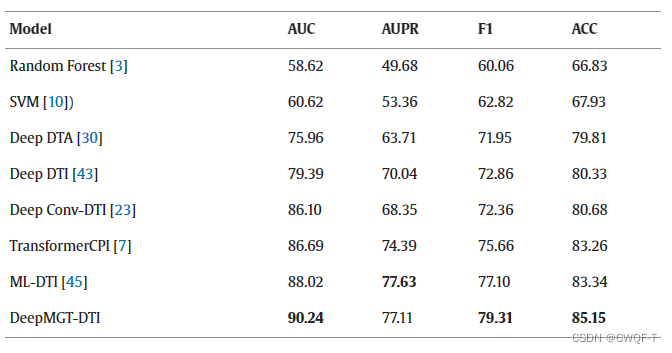
此外,作者还预测了抑制COVID-19的Delta变体的药物作为测试,在此不赘述。
三.GCHN-DTI: Predicting drug-target interactions by graph convolution on heterogeneous networks
通过异构网络上的图卷积预测药物-靶点相互作用 2022.10 二区
1.模型
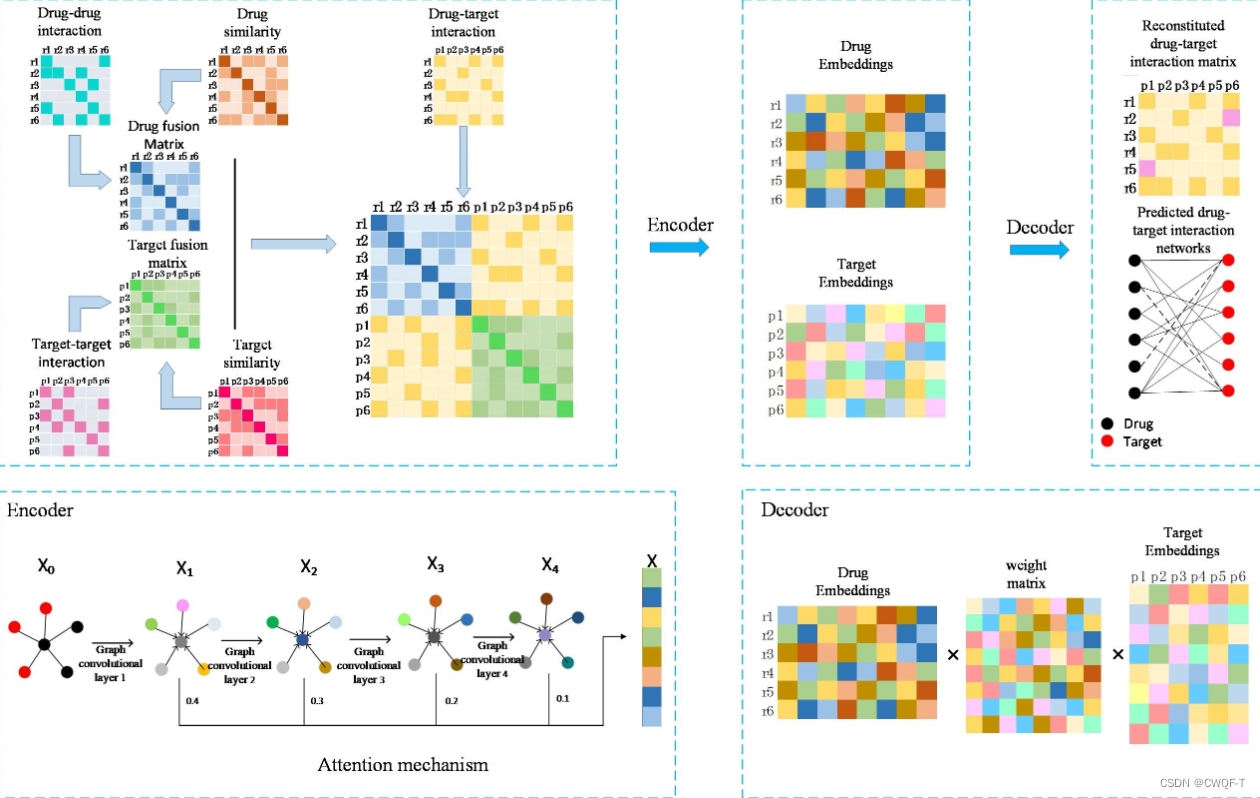
GCHN-DTI整合了药物-靶点相互作用、药物-药物相互作用、药物相似性、靶点-靶点相互作用和靶点-相似性的网络信息。在异构网络中采用图卷积运算,得到药物和靶点的节点嵌入;此外,我们在图卷积层之间加入了注意力机制,以组合来自每一层的节点嵌入。最后预测。
1.将药物网络和靶点网络通过线性组合进行融合,得到药物融合网络和靶点融合网络,然后将药物融合网络、靶点融合网络和药物-靶点相互作用网络构建为异构网络。药物融合网络是由药物-药物相互作用以及药物相似性矩阵融合而成;靶点融合网络是由靶点-靶点相互作用以及靶点相似性矩阵融合而成。
2.将包含药物信息和蛋白质信息的异构网络馈入图卷积网络的编码器中,得到药物和蛋白质的低维表示。
3.将可训练权重矩阵、药物低维矩阵和蛋白质低维矩阵输入解码器,得到重构的相互作用矩阵。相互作用矩阵的每个位置都代表一个新的药物-靶点相互作用分数。输入到预测网络中进行预测。
1.药物表示
SMILES
2.蛋白质表示
序列
2.实验
1.数据集
蛋白质-蛋白质相互作用从HPRD数据库
药物化学结构相似性网络中,使用Tanimoto系数计算一对两种药物之间的相似性得分。在靶蛋白序列相似性网络中使用基于其主要序列的Smith-Waterman 评分计算一对蛋白质之间的相似性评分。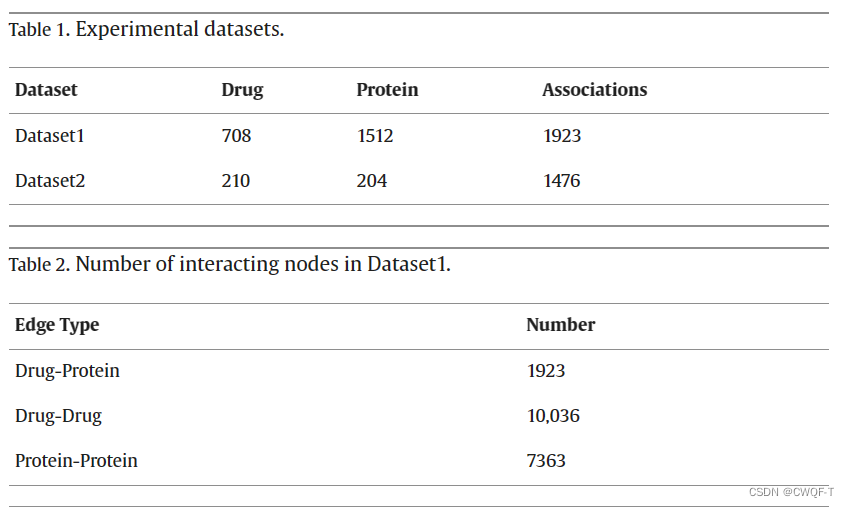
3.结果
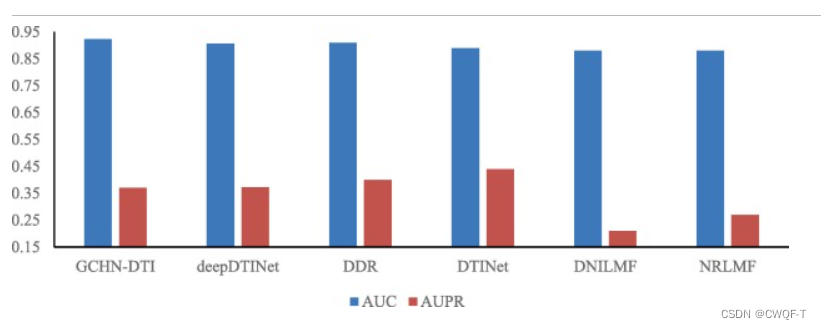
消融实验:
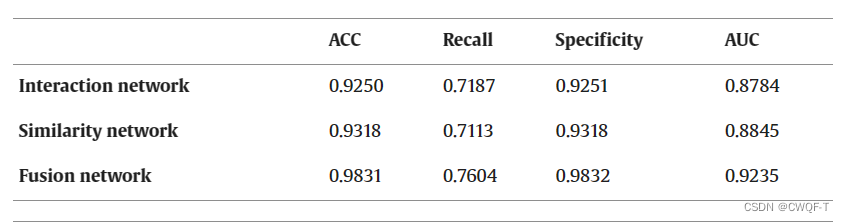
四.BACPI: a bi-directional attention neural network for compound–protein interaction and binding affinity prediction
用于化合物-蛋白质相互作用和结合亲和力预测的双向注意力神经网络 2022.3 一区
1.模型
SMILES使用RDKit将化合物转换为图形,表示为原子邻接图,蛋白质表示为氨基酸序列,分为不同的子序列,这些子序列被视为残基。分别使用 GAT 和 CNN 从原子结构图和残基序列中来学习原子和氨基酸的表示。最后将这些特征输入双向注意力神经网络架构,以整合化合物和蛋白质的表示,可以引导模型关注原子和氨基酸的局部有效位点,从而增加模型的可解释性。最后,将化合物-蛋白质对的集成向量表示输入分类器以预测 CPI 或回归模型以预测化合物-蛋白质结合亲和力。
药物:第 j个顶点特征对第 i个顶点的重要性计算:||是串联的意思
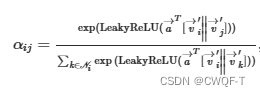
通过结合K个独立的图注意力层的输出特征,将该机制扩展到多头注意力。再通过一层网络进行变化到复合空间,最终得到一个原子特征。还使用了ECFP扩展指纹,得到长为1024的特征向量长度,最后通过多层神经网络转化为复合空间。
蛋白质:3-gram进行处理(每次移动一个氨基酸来生成一堆子序列),把得到的子序列认为是“残基”。把残基转化为随机初始化的嵌入,并且通过CNN进行更新,得到最终的输出。
过程:把得到的指纹特征、原子特征、残基特征转化为相同维度.得到蛋白质矩阵P和化合物矩阵C(好像没有加上指纹特征?)。通过计算A=tanh(CUPt)得到交互矩阵,每个值表示交互强度,U是可训练的参数矩阵。下面式子中的Ic是从残基传递给原子的信息;Ip是从原子传到残基的信息。

双向注意力α计算:a2r是从原子到氨基酸残基的注意力

最后hc=αa2r⋅C是原子特征的加权总和,hp=αr2a⋅P是蛋白质特征的加权总和。
通过执行L次上述过程,会得到不同的蛋白质特征和化合物特征,串联这些特征得到最终的蛋白质特征和化合物特征,再计算交互作用。最后一步计算交互作用的时候才加上指纹特征
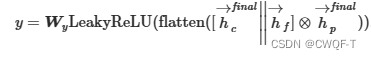
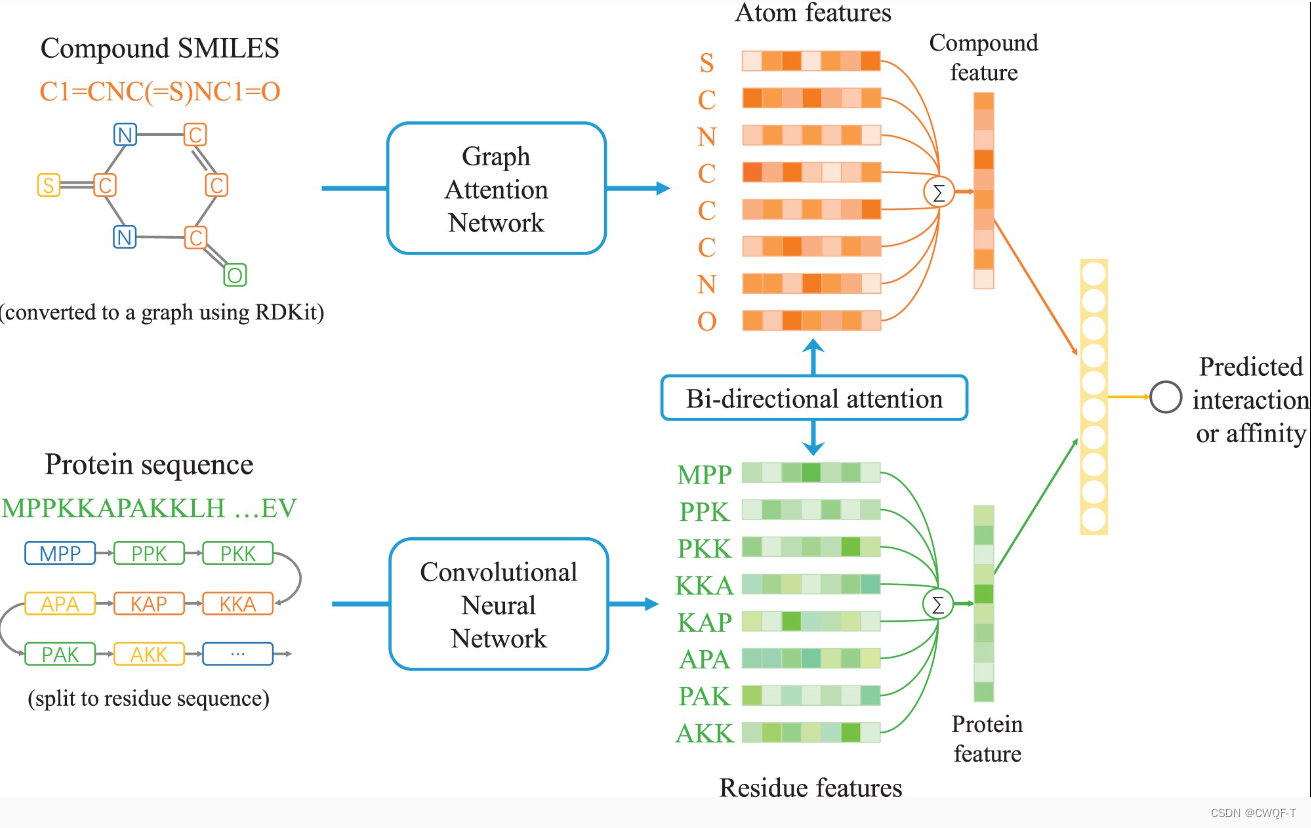
1.药物表示
根据SMILES得到原子邻接图和指纹
2.蛋白质表示
序列
2.实验
1.数据集
1.CPI实验
使用Improving compound–protein interaction prediction by building up highly credible negative samples论文的human和celegans数据集.五重交叉验证
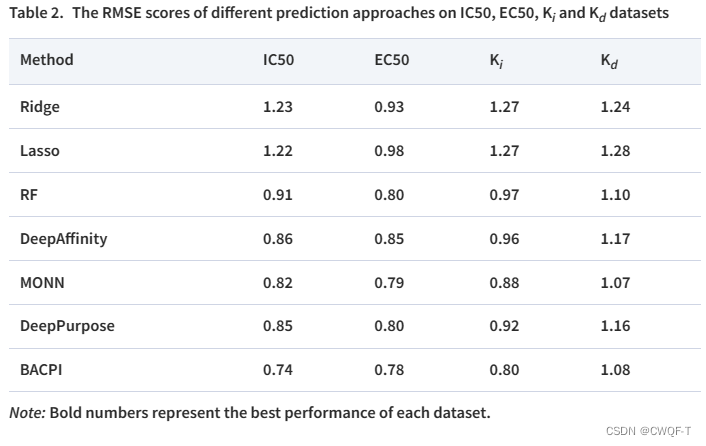
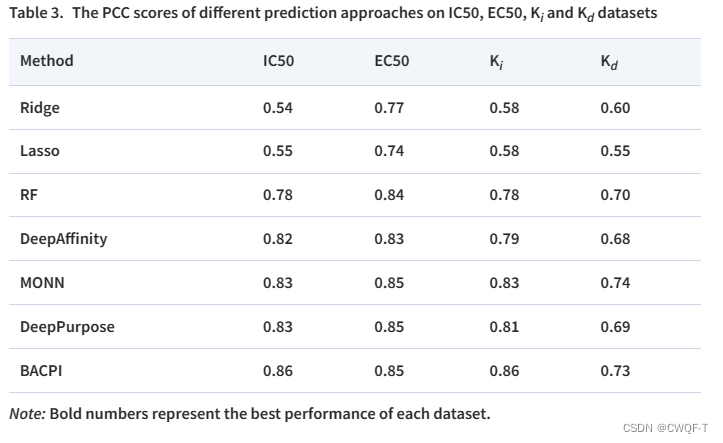
2.亲和力实验
使用论文DeepAffinity: interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks.的BindingDB数据集

3.结果
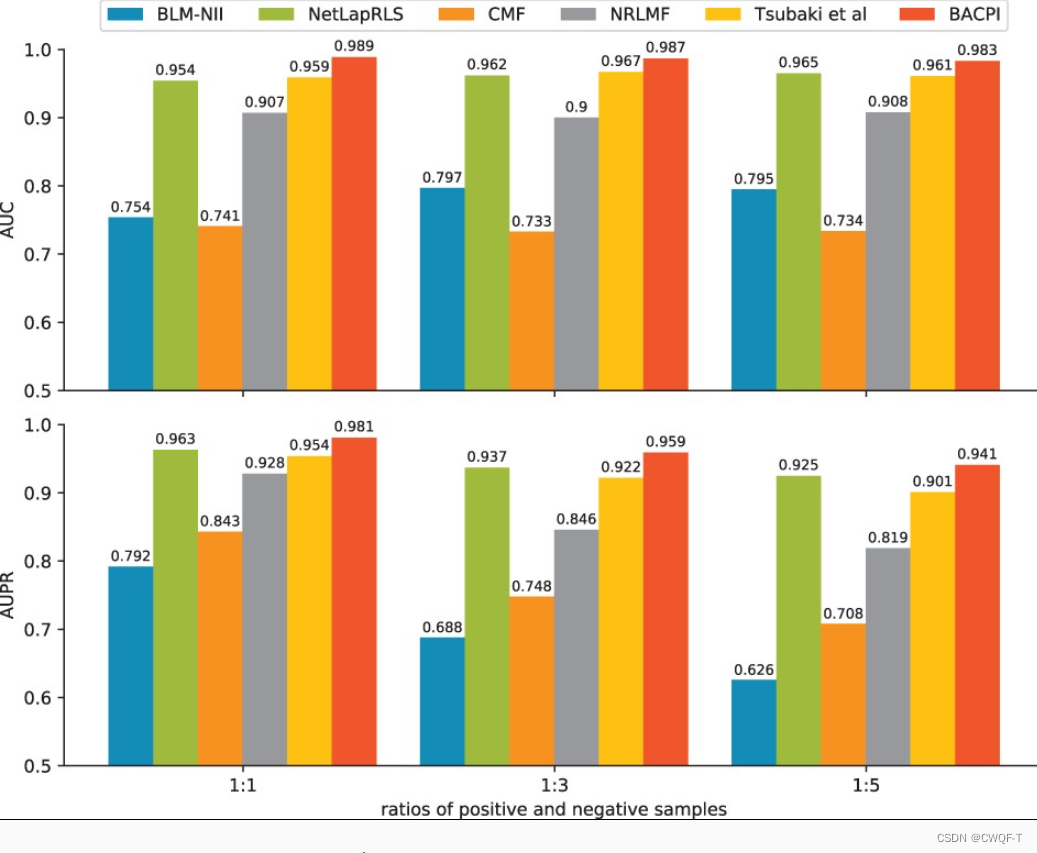
在处理不平衡的数据集时,AUPR 可以更准确地评估性能,而 AUC 对性能的评价比较乐观。
1.CPI
human
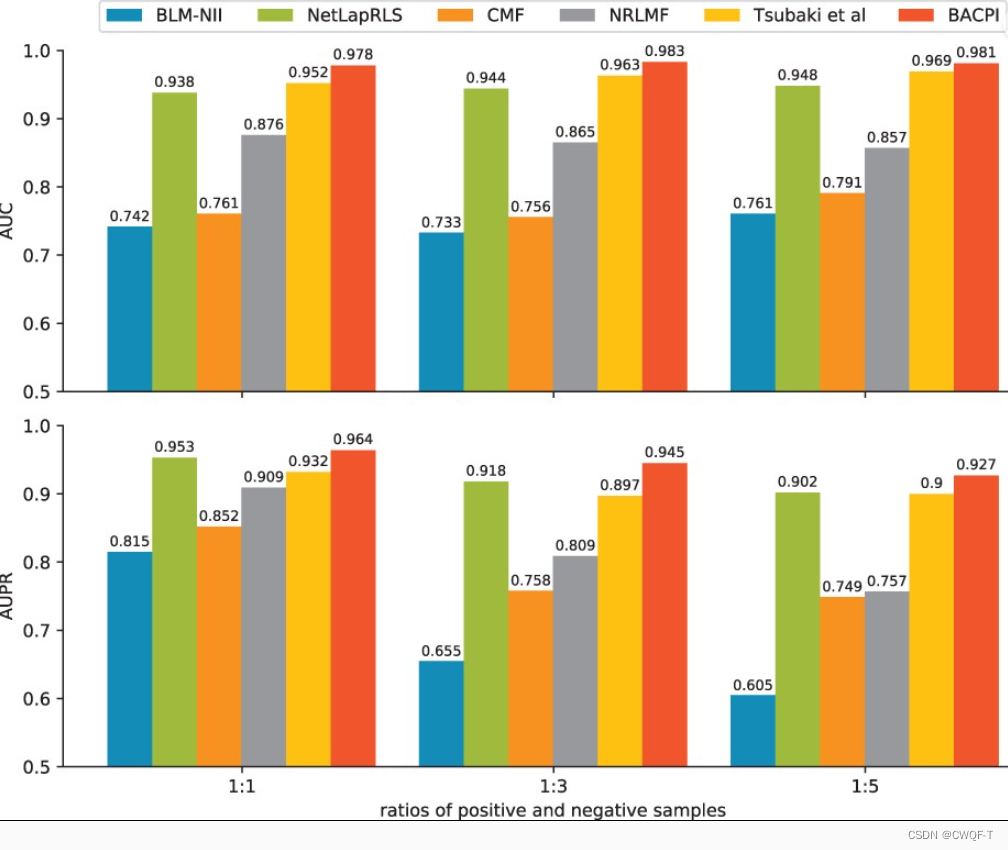
celegans
2.亲和力

















 566
566

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









