






一.MDL-CPI: Multi-view deep learning model for compound-protein interaction prediction
用于化合物-蛋白质相互作用预测的多视角深度学习模型 二区 2022.08
1.模型
利用BERT和CNN从顺序角度提取蛋白质特征,使用GNN从结构角度提取复合特征,并使用AE2网络生成统一的特征空间,以学习BERT-CNN 和 Graph 嵌入。
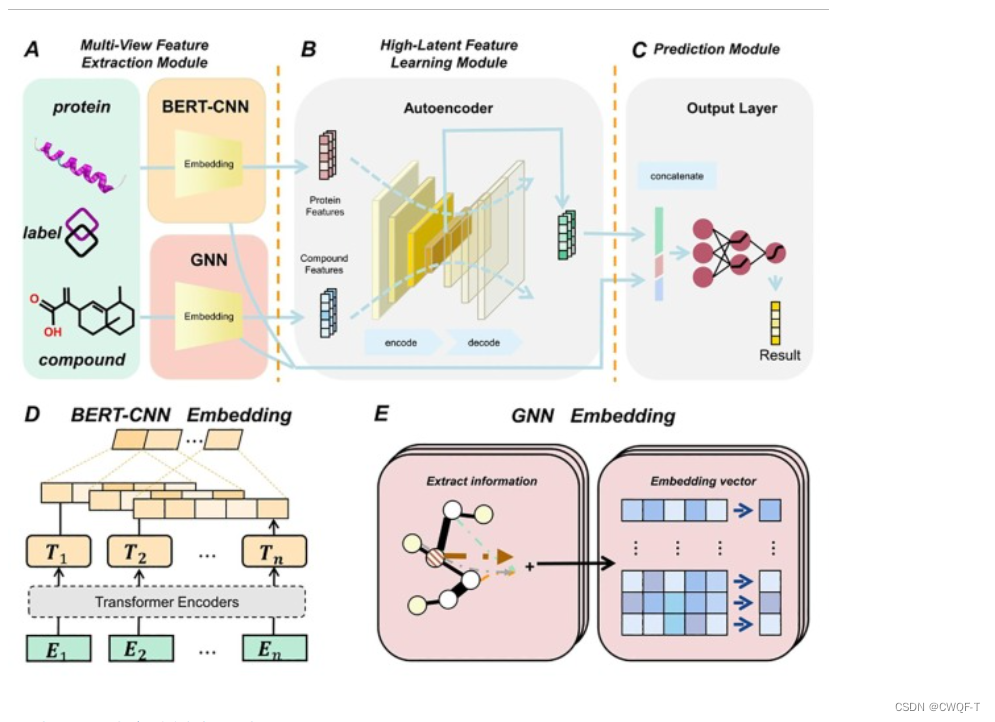
蛋白质:
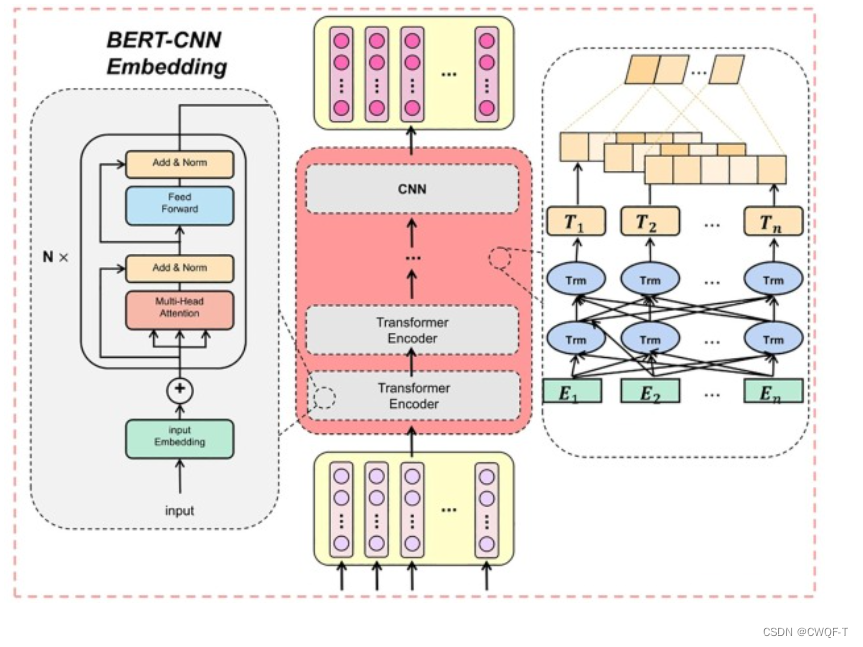
先将一个蛋白质序列拆分为重叠的 3 -gram氨基酸序列,再输入到BERT中。BERT的编码器的输出作为整个BERT的输出,再输入到CNN中获取低维特征。
药物:得到每个smiles的边索引矩阵和邻接矩阵(都是Natom*Natom大小)。再通过一维Weisfeiler-Lehman算法(1-WL)提取了它的指纹,通过one-hot编码得到指纹矩阵,送到GNN得到药物的结构特征。
AE2 由 AE-net(内部 AE 网络)和DG-net(退化网络)组成,前者从每个视图中提取特征,后者将完整的潜在表示映射回原始特征。蛋白质特征和化合物特征分别通过AE-net和DG-net。AE-net(内部AE网络)用于从每个视图中提取内在特征,DG-net(降解网络)用于学习可用于重建每个视图的潜在多视图表示。
将蛋白质特征、化合物特征和统一特征信息进行组合,并将组合后的特征发送到预测模块中
1.药物表示
SMILES通过GNN
2.蛋白质表示
氨基酸序列通过BERT和CNN
2.实验
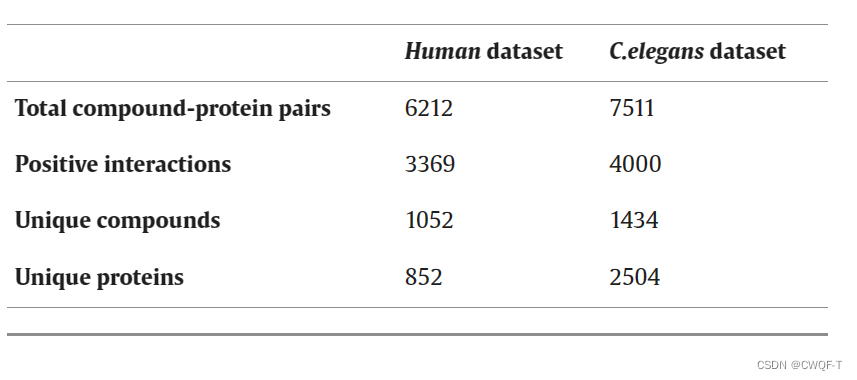
1.数据集
3.结果
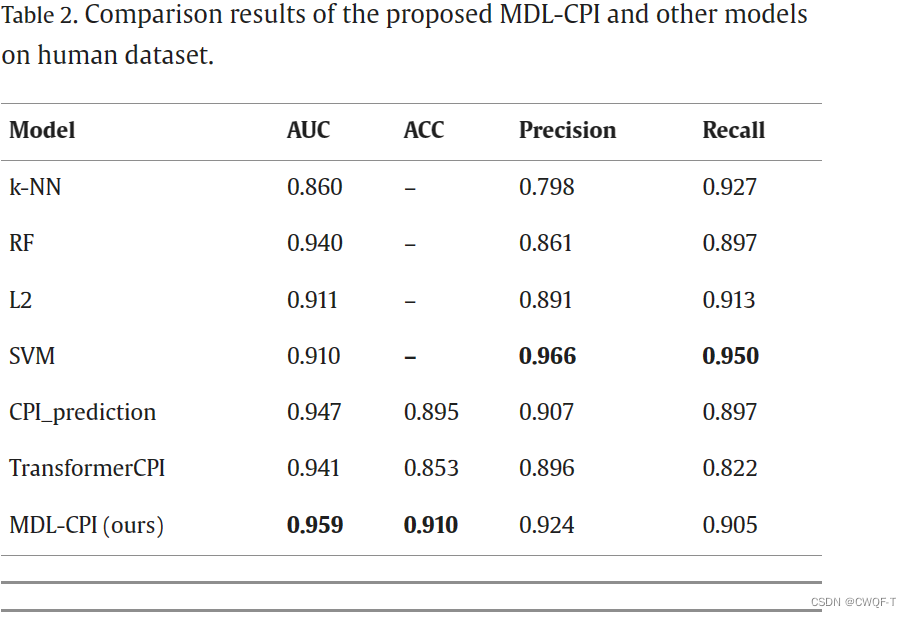
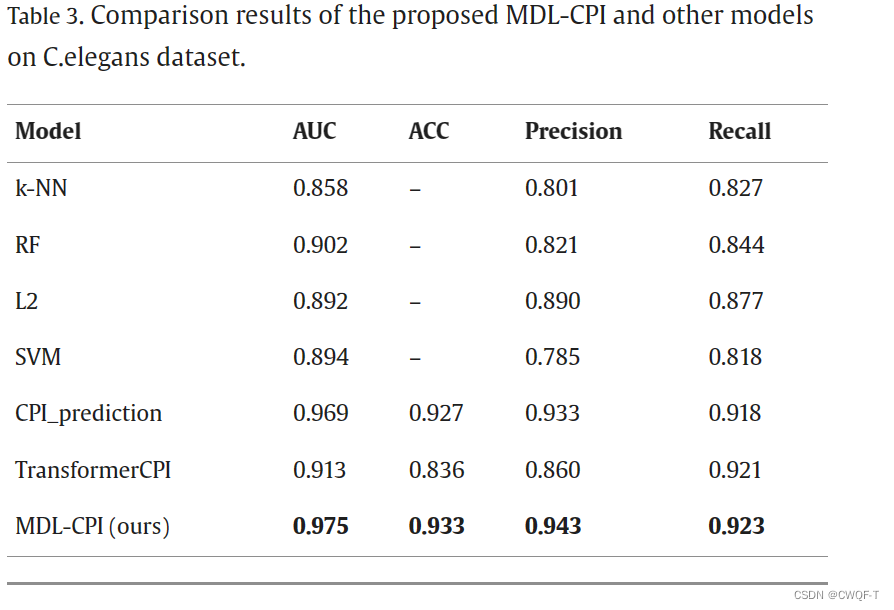
消融实验:仅使用BERT或者CNN提取蛋白质特征
可以看到只是用一种来提取特征时候,鲁棒性不够,性能波动大。两者都使用的时候可以捕获蛋白质的全局和局部信息,增大了鲁棒性。
二.DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences
通过对蛋白质序列进行卷积的深度学习预测药物-靶点相互作用 2019.06 生信二区
作者提出:对于蛋白质,通常使用组成、过渡和分布(CTD)描述符作为计算表示。但是,使用蛋白质描述符和药物指纹图谱的基于特征的模型比以前的常规定量构效关系(QSAR)模型表现出更差的性能
1.模型
用基于CNN的深度学习方法 检测靶蛋白局部残基模式的预测模型成功地丰富了原始蛋白质序列的蛋白质特征。
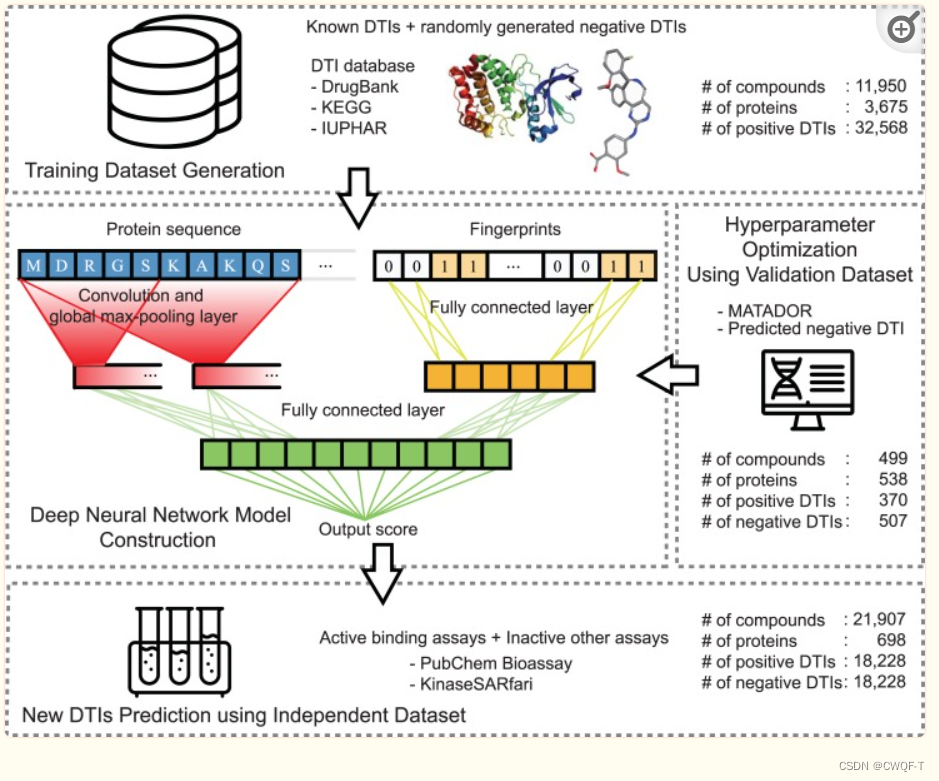
训练模型:使用从各种DTI数据库中收集的大量DTI数据(DrugBank、KEGG、IUPHAR)
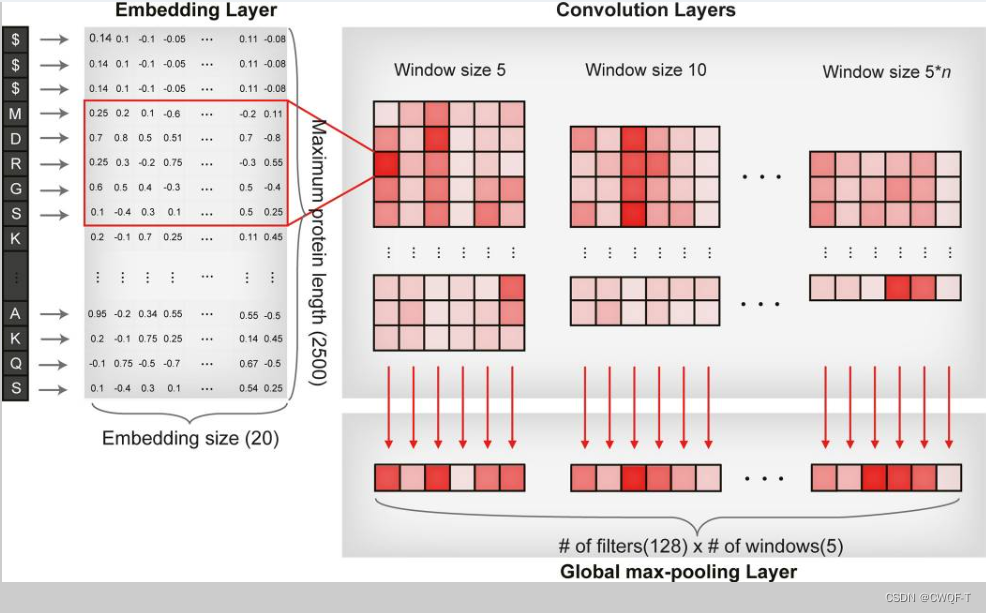
构建模型:把整个蛋白质序列转化为固定大小的embedding向量,再采用CNN来捕获局部残基(参与DTI作用的),每个卷积结果都进行最大池化。最连接最大池化的结果作为后续模型的输入向量,最后得到蛋白质特征。
蛋白质特征与 从全连接层得到的药物的摩根指纹(而不是扩展连接指纹(ECFP))中 得到的药物特征连接起来,使用全连接层来预测DTI。最后使用MATADOR的DTI和Liu等人预测的负相互作用优化了模型。(??)
1.药物表示
SMILES
2.蛋白质表示
蛋白质序列
2.实验
1.数据集
PubChem和KinaseSARfari数据集
3.结果
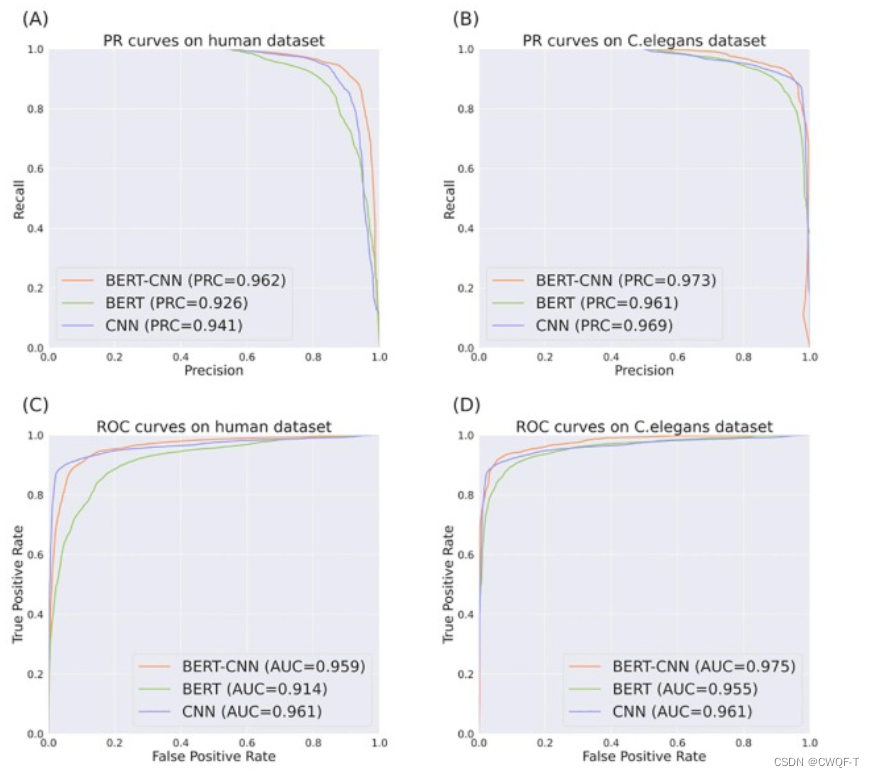
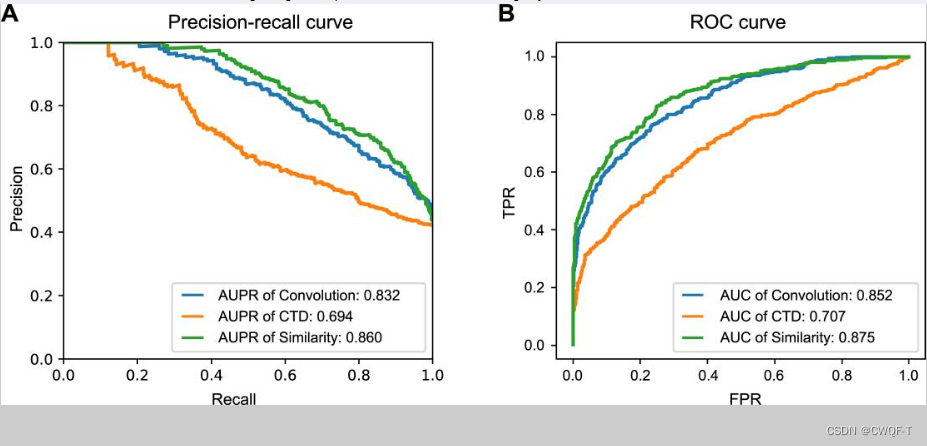
1.与其他蛋白质的表示方法相比:蛋白质使用CTD描述符方法表示或者使用相似性矩阵表示。
使用相似性描述符的KinaseSARfari数据集的模型性能与所提出的模型相似,作者解释为相似性描述符作为结构域水平的局部残基模式而不是整个蛋白质复合物的信息特征。而CTD 的信息量和丰富度低于其他描述符
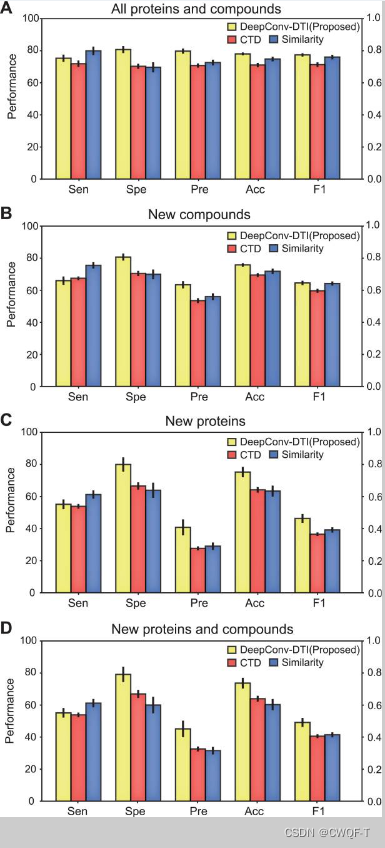
2.分类:
(A) 所有查询的 PubChem 数据集。 (B) PubChem 数据集,其化合物不在训练数据集中。(C) 目标不在训练数据集中的 PubChem 数据集。(D) 化合物和靶点不在训练数据集中的 PubChem 数据集
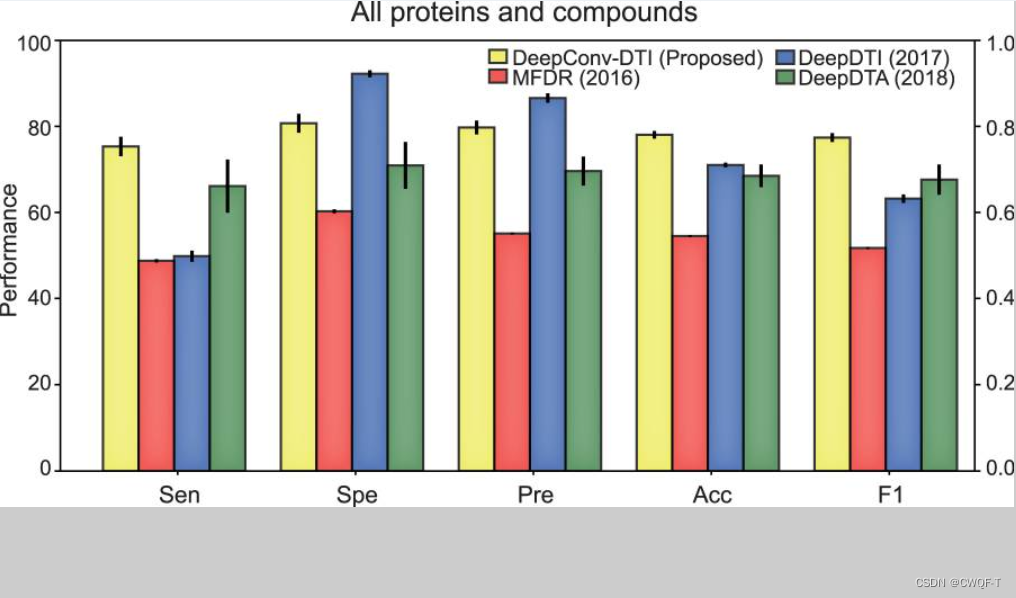
3.与其他模型比较:
三.MGraphDTA: deep multiscale graph neural network for explainable drug–target binding affinity prediction
用于可解释药物-靶标结合亲和力预测的深度多尺度图神经网络 2022 二区
问题:作者认为认为层数较少的GNN不足以捕获化合物的全局结构,而且堆叠深层GNN会导致梯度消失和过渡平滑问题(即随着模型深度的增加,越来越多的原子被包含在某个顶点的表示中,导致两个原子 C2 和 C1 的顶点嵌入变得越来越相似);并且应该保持化合物的局部结构,图注意力机制只考虑一个顶点的领域,无法捕捉每个分子的原子之间的全局关系。
1.模型
多尺度神经网络MGNN和用于DTA解释和预测的梯度加权亲和激活映射(Grad-AAM)(一种可视化方法???)
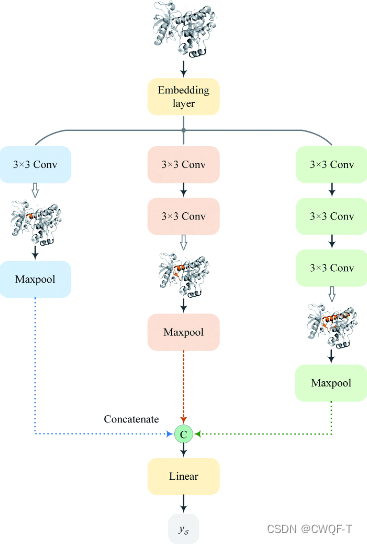
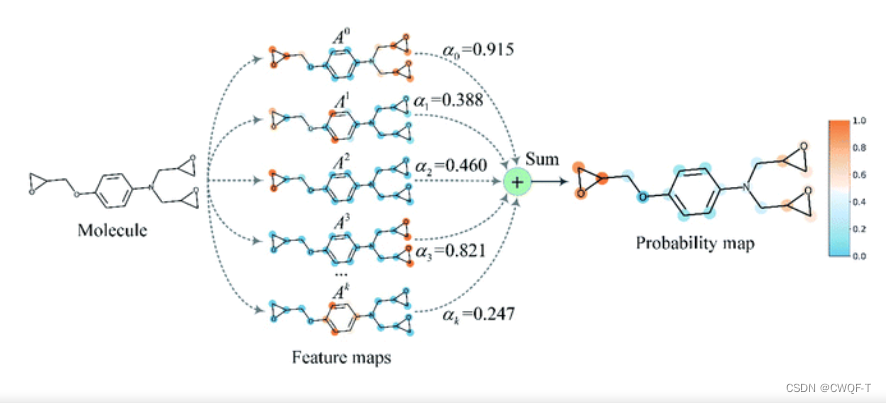
采用具有27个图卷积层的MGNN和多尺度卷积神经网络(MCNN)分别提取药物和靶点的多尺度特征。该药物的多尺度特征包含有关不同尺度分子结构的丰富信息,使GNN能够做出更准确的预测。将提取的药物和靶标的多尺度特征分别融合,然后串联以获得给定药物-靶标对的组合描述符。将组合描述符输入MLP以预测结合亲和力。Grad-AAM 使用流入 MGNN 最终图卷积层的亲和力梯度来生成概率图,突出显示对 DTA 贡献最大的重要原子。
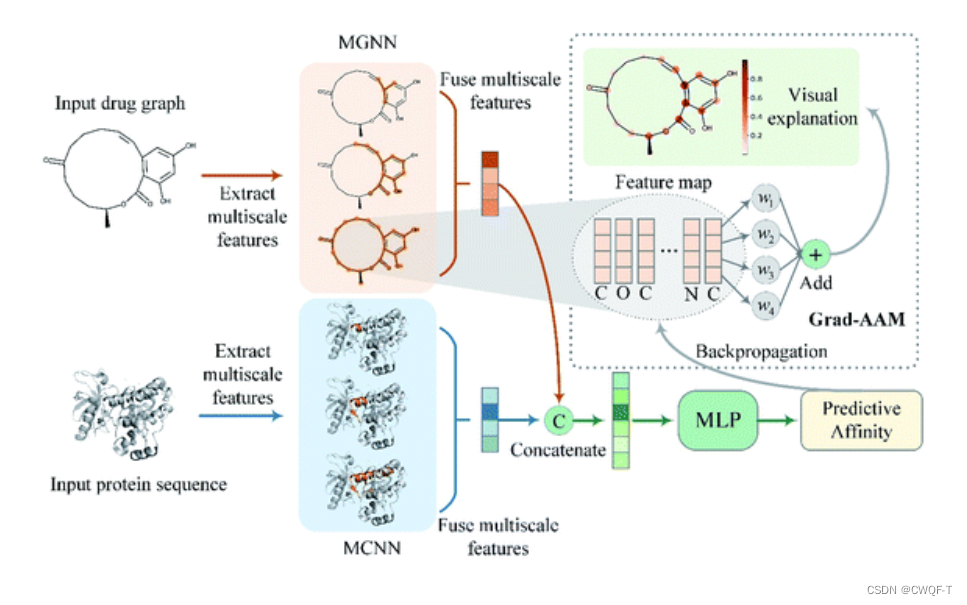
MGNN:
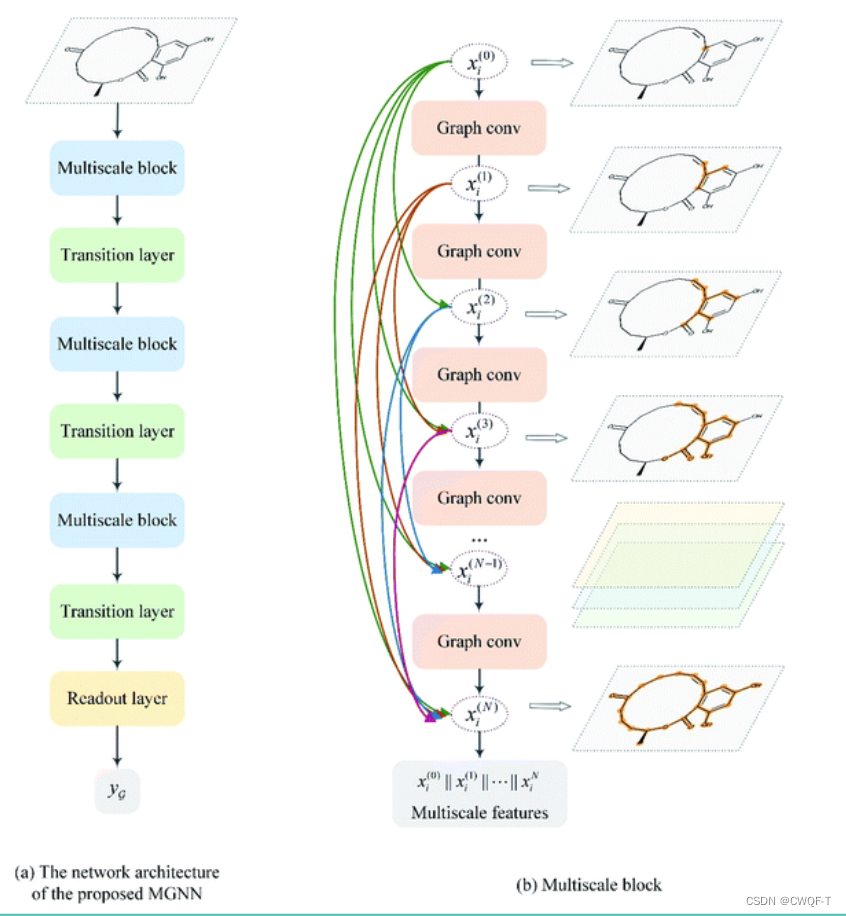
密集连接以前馈方式将每一层连接到其他每一层,密集连接允许所有层直接访问以接收相对于每个权重的损失函数的梯度,从而避免梯度消失问题,并允许训练非常深的 GNN。 n 表示第 n 层GNN,∥ 是串联运算,H是由W1,W2组成,那经过多层GNN处理之后的Xi表示为:
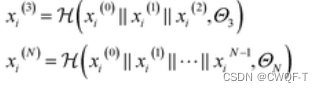
其中每个GNN 都使用如下公式得到Xi。
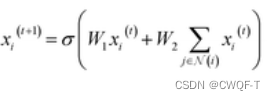
这样就得到了分子在局部和全局环境中的结构信息。
对于每两个多尺度模块之间,添加的过渡层旨在整合先前多尺度块中的多尺度特征,并减少特征图的通道数到之前的一半。过渡层如下: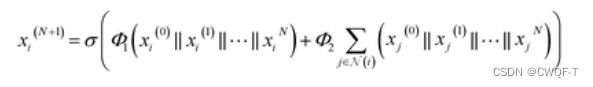
MCNN:设计了一个由堆叠3*3卷积层组成的具有不同感受野(感受野=(前一层输出维度-1)*步长+卷积核大小)的三个分支的网络,以检测不同尺度的蛋白质的局部残基模式。三个感受野分别是3,5,7。S是1200*128(嵌入向量大小)大小。F是三个分支,m是最大池化。
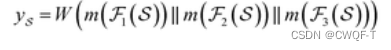
Grad-AAM:使用流入 MGNN 最后一个图卷积层的梯度信息来了解每个神经元对亲和力决策的重要性:将最后一个图卷积层的特征图表示为A,为了获得给定分子的顶点数的化学概率图,首先计算了亲和力评分P相对于神经元A的梯度,然后计算通道重要性权重:
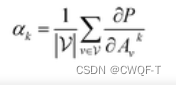
最后,采用最小-最大归一化对概率图PGrad-AAM映射到0-1
1.药物表示
SMILES,处理为分子图
2.蛋白质表示
序列(构建词汇表,将氨基酸序列映射为整数序列,最大长度固定为1200,嵌入为128维向量)
2.实验
1.数据集
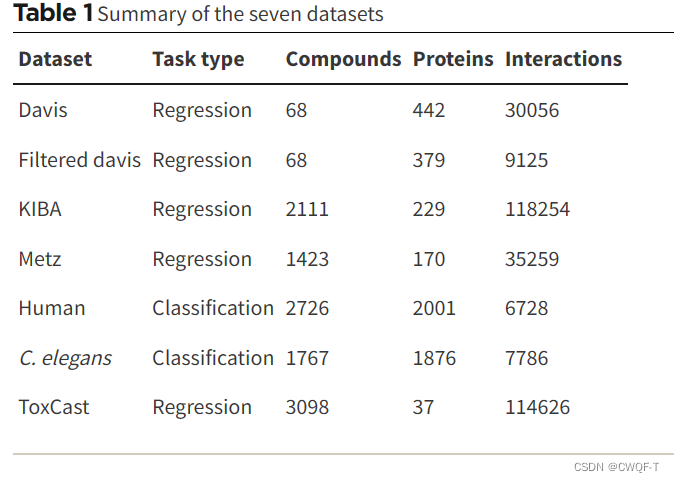
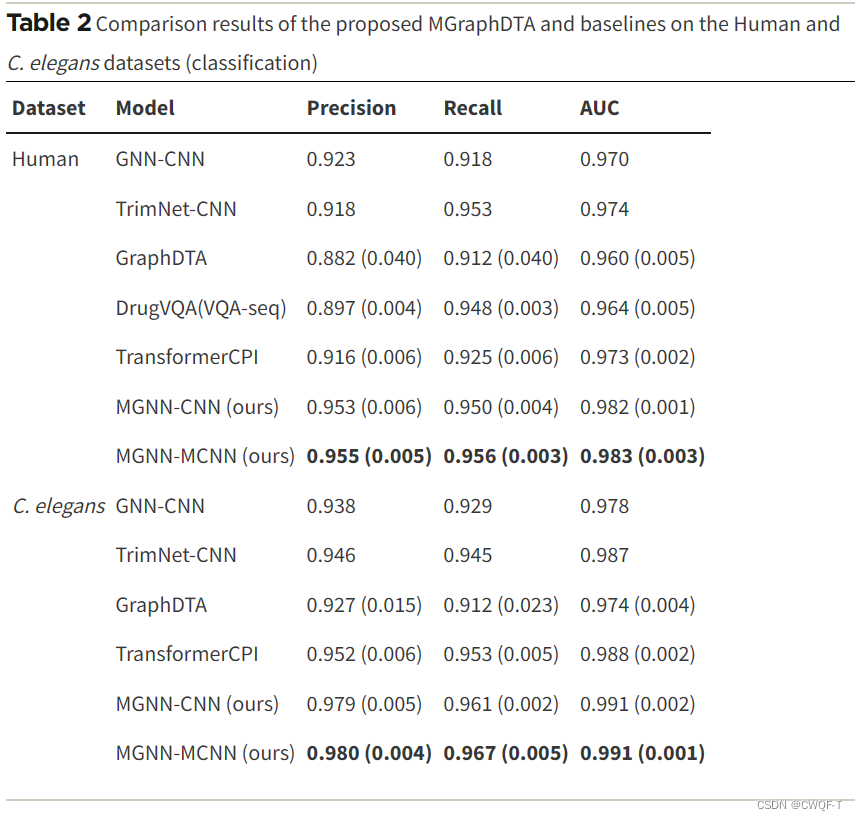
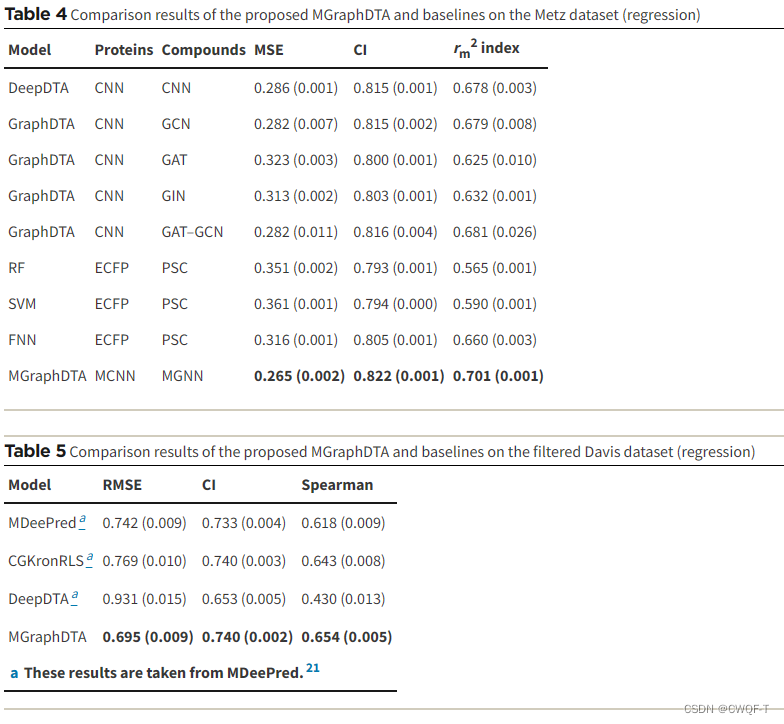
3.结果
分类任务中:
回归任务中:
四.GraphATT-DTA: Attention-Based Novel Representation of Interaction to Predict Drug-Target Binding Affinity
用于预测药物-靶标结合亲和力和区域的多功能稳健模型 2023.2 二区
问题:在提取丰富的蛋白质和药物特征方面存在不足。蛋白质或药物的元素,如氨基酸和原子,对预测任务有重大影响。1D 卷积和 MLP 都完全忽略了每个元素的单独特征;LSTM和GNN直接提取单个特征,但不足以获取全局特征;2D 卷积通过增加卷积核或堆叠更多卷积层来提取单个和全局特征,但其计算消耗迅速增长;基于Transformer的骨架在参数上过于冗余,使其对高效的蛋白质和药物特征提取不太友好。另一方面,很多基于注意力机制的方法试图通过高注意力响应来识别BR(交互结合区域),这缺乏理论依据。因为突出显示的区域与蛋白质的生物学特性无关(?相关吧)。
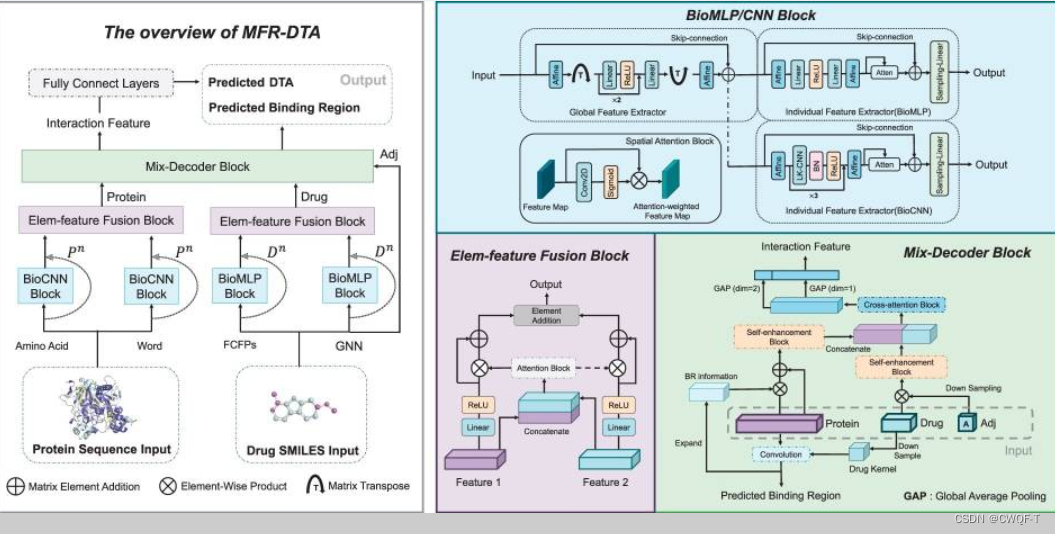
1.模型
1.设计了一种新的生物序列特征提取模块,即BioMLP,它帮助模型提取序列元素的单个特征(使用空间注意力块ATT())和全局特征。
蛋白质特征:使用CNN提取。包括全局和单个特征(氨基酸特征)
药物:使用FCFP和GNN特征。使用MLP提取。包括全局和单个特征(原子特征)
全局特征使用相同的架构提取:ResMLP(AF块)+3个全连接层+Relu
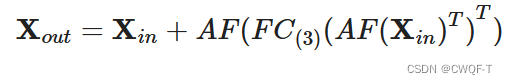
单个特征的提取器是不同的:在BioMLP中,FC包含两个全连接层和一个 ReLU 函数层。在BioCNN中,CNN包含三个卷积块(对不同的数据集、同一数据集的不同卷积块都是不一样的),每个卷积块由一个一维卷积层、一个批量归一化层和一个 ReLU 层组成。此外,特征提取块中的空间注意力模块ATT(),通过二维卷积CNN2D捕获相邻元素之间的局部关系并通过 Sigmoid 函数对捕获的信息进行归一化
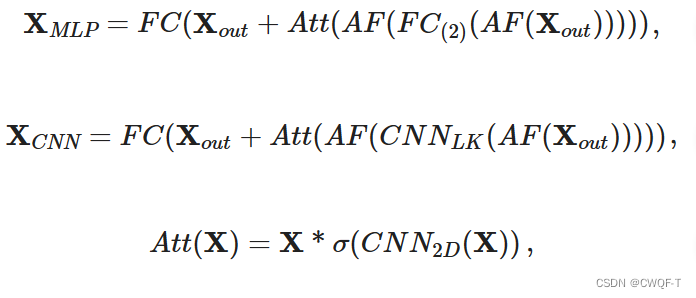
2.Elem特征融合块通过组合单个和全局两种类型的特征来生成注意力矩阵,以确保突出显示每种类型特征的互补元素。
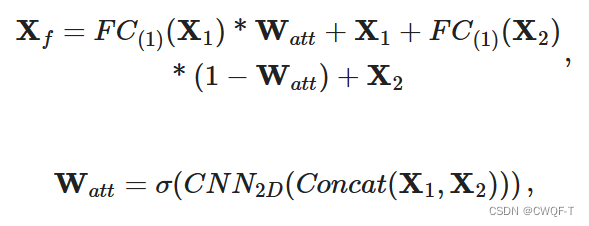
3.构建了一个 Mix-Decoder 模块,该模块提取药物-靶标相互作用信息并同时预测它们的结合区域。输入是蛋白质特征向量、药物特征向量和药物邻接矩阵。
(1)得到结合区域:通过使用线性层采样药物来得到药物核,再通过卷积药物核与蛋白质特征来得到响应向量S。在响应向量中,具有最高值的元素被识别为药物靶标响应区域BR。
(2)得到DTI: 两个S-E模块:一个将反应向量经过重复填充来扩展成为大小为Lp×C的信息矩阵BRinformation,再与蛋白质特征进行元素乘法,以鼓励模型专注于 BR;再与本来的蛋白质向量相加。另一个将邻接矩阵通过下采样(?)得到原子连通向量,并且通过重复填充扩展为邻接信息矩阵M,再通过逐元素地将M与药物特征Xd进行乘法运算,可以将药物特征Xd突出显示在特征聚合区域上。 两个S-E模块得到的向量相连接,再通过交互注意力模块来提取DTI特征,得到的输出矩阵再在两个维度上分别进行全剧平均池化,得到的两个向量串联起来得到最终的交互特征。
1.药物表示
SMILES
2.蛋白质表示
序列
2.实验
1.数据集
KIBA 和 Davis ,用3D数据集sc-PDB评估BR预测方法的性能
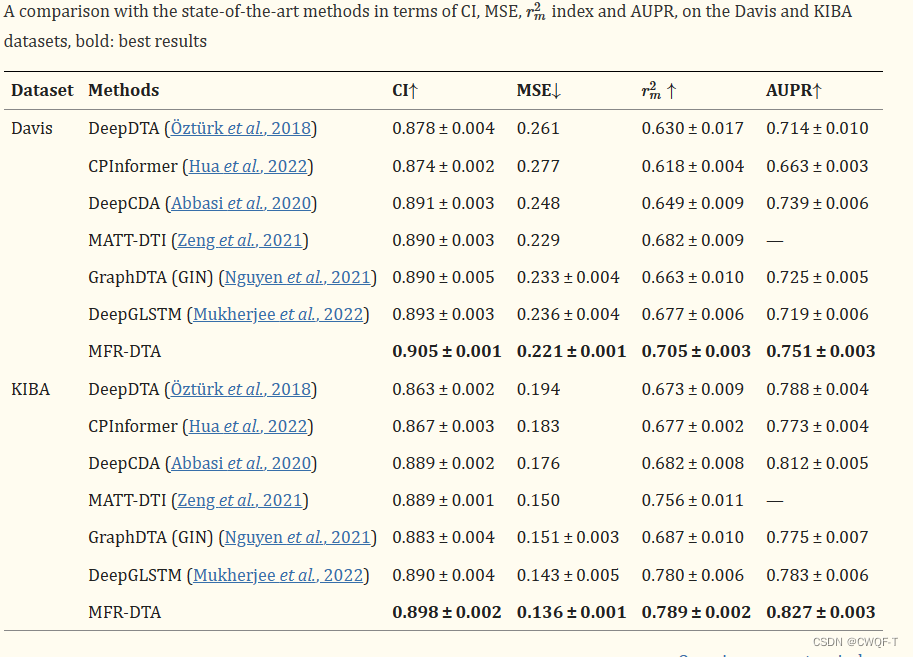
3.结果
注意:使用了 Uniprot数据集中收集所有蛋白质的结合位点信息,并将它们嵌入为标记载体,大小为LP×1.在标签嵌入中,将非 BR 中的元素编码为 0,将 BR 中的元素编码为亲和值。并且采用整流翼(RWing)损失函数(没听过,DTI中没见过)。

















 2032
2032

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









