







引言
在现代化学和生物信息学中,分子表示方式是进行分子建模、药物设计和生物大分子研究的重要工具。这些表示方式能够将分子的结构信息简化为可处理的形式,便于计算机程序读取、存储和操作。在诸多分子表示方式中,SDF(Structure Data File)、Mol2和SMILES(Simplified Molecular Input Line Entry System)是最常用的几种格式。
SDF与Mol2的重要性
SDF文件是一种广泛用于化学和生物领域的分子文件格式,特别是在三维分子建模和分子对接任务中。SDF格式不仅能表示分子的连接方式,还能记录原子的空间位置、键的类型、以及其他分子属性。这种详细的结构信息使得SDF文件在药物开发、分子动力学模拟和三维可视化等方面有着重要应用。以下展示的是一个典型的SDF文件的格式及其对应的分子图示:
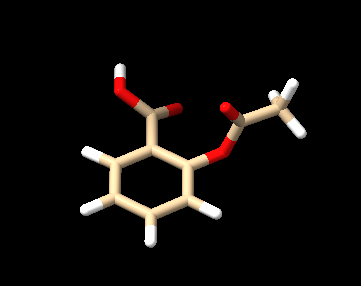
与SDF文件类似,Mol2文件也常用于分子的三维结构表示。Mol2文件除了可以记录分子结构和坐标信息外,还可以表示多种分子类型的属性,例如蛋白质、配体、药物小分子等。Mol2文件特别适合在药物设计软件中进行分子模拟和配体对接。
SMILES的重要性
与SDF和Mol2文件不同,SMILES通过简单的字符串来描述分子的拓扑结构。由于其紧凑、易读的格式,SMILES广泛应用于分子库的存储、搜索、以及药物开发中的虚拟筛选。以下是一个典型的SMILES表示及其对应的分子结构图:
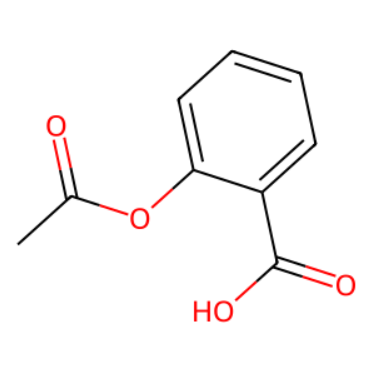
这个SMILES描述了该分子的结构:
- CC(=O)O 代表一个乙酰基(acetyl group),位于分子左侧。
- C1=CC=CC=C1 代表一个苯环(benzene ring),位于中间部分。
- C(=O)O 代表一个羧酸基(carboxyl group),位于右侧。
尽管SMILES比SDF和Mol2文件更简洁,但它在记录分子三维结构时较为有限。因此,SMILES更适用于快速搜索和化合物库的管理,而SDF和Mol2文件则更适合三维结构的精确模拟。
接下来,我们将探讨如何从PDB文件中提取分子的SDF和Mol2文件,以及如何从这些文件中获取分子的SMILES表示,并分析这些格式在实际应用中的优劣。
1 通过PDB文件提取配体分子
示例
7A0C:RCSB PDB - 7A0C: X-ray structure of NikA from Escherichia coli in complex with Fe-6-Me2-BPMCN
PDB文件不仅包含蛋白质的三维结构信息,还可能包含与蛋白质结合的小分子,例如药物分子、辅因子、金属离子等。这些分子通常记录在PDB文件的HETATM部分。我们可以通过Biopython 和 OpenBabel提取指定名称的配体分子,并将它们分别保存为SDF文件或MOL2文件。
1.1 PDB文件中的配体信息
在PDB文件中,配体分子通常记录在HETATM部分,标识符为配体的残基名(Residue Name),例如“QTT”表示特定的配体分子。每个配体分子可能出现在多个链(Chain)中,且可能存在多个副本。因此,在提取配体时,需要指定残基名,并且可以根据链ID进一步区分。
示例:
HETATM 8010 N1 QTT A 602 1.772 -2.375 34.336 1.00 41.23 N
HETATM 8011 C1 QTT A 602 2.186 -1.989 35.672 1.00 38.26 C
HETATM 8012 O1 QTT A 602 5.279 -5.886 36.039 1.00 25.03 O
HETATM 8013 O2 QTT A 602 3.249 -5.090 35.931 1.00 23.99 O
HETATM 8014 C2 QTT A 602 -1.852 -4.250 31.505 1.00 52.42 C
HETATM 8015 C3 QTT A 602 0.330 -8.303 32.862 1.00 33.42 C
...
HETATM 8106 N1 QTT B 601 34.781 20.935 19.575 1.00 37.90 N
HETATM 8107 C1 QTT B 601 35.482 19.762 19.090 1.00 35.53 C
HETATM 8108 O1 QTT B 601 37.688 18.893 23.490 1.00 21.45 O
HETATM 8109 O2 QTT B 601 37.423 20.039 21.649 1.00 21.87 O
HETATM 8110 C2 QTT B 601 33.980 25.917 19.260 1.00 53.52 C
...
该示例显示了QTT配体的多个原子,分布在A链和B链中,每个链上都有一个编号为602或601的配体。
1.2 使用Biopython提取特定配体并保存为SDF
我们可以使用Biopython 从PDB文件中提取特定名称的配体(例如QTT),并将其保存为单独的PDB文件。虽然Biopython本身不支持直接导出为SDF文件,但可以结合OpenBabel 或 RDKit 进行转换。
以下代码展示了如何使用Biopython提取指定名称的配体:
from Bio.PDB import PDBParser, PDBIO, Structure, Model, Chain, is_aa
# 初始化PDB解析器
parser = PDBParser(PERMISSIVE=1)
structure = parser.get_structure("PDB_ID", "../../data/7A0C/7a0c.pdb")
# 创建一个PDBIO对象用于保存
io = PDBIO()
# 创建一个列表存储所有符合条件的配体
ligands = []
# 遍历结构中的所有链,提取特定名称的配体(例如 QTT)
for model in structure:
for chain in model:
for residue in chain:
if residue.get_resname() == "QTT" and not is_aa(residue):
ligands.append((residue, chain))
# 保存提取的每个配体为单独的PDB文件
for idx, (ligand, chain) in enumerate(ligands):
# 创建一个新的结构,用于保存单个配体
new_structure = Structure.Structure(f"LIG_{idx + 1}")
new_model = Model.Model(0) # 单模型
new_chain = Chain.Chain(chain.get_id()) # 使用原始链ID
new_chain.add(ligand)
new_model.add(new_chain)
new_structure.add(new_model)
# 设置新的结构并保存
ligand_id = ligand.get_resname() + "_" + str(ligand.get_id()[1]) + "_" + chain.get_id()
io.set_structure(new_structure)
output_pdb_file = f"ligand_{idx + 1}_{ligand_id}.pdb"
io.save(output_pdb_file)
print(f"Saved {ligand_id} to {output_pdb_file}")
执行上述代码后,将生成文件ligand_1_QTT_602_A.pdb 和 ligand_2_QTT_601_B.pdb。可以手动检查这些文件中的配体部分是否与原PDB文件保持一致。
例如,提取的ligand_1_QTT_602_A.pdb的开头和结尾:
HETATM 1 N1 QTT A 602 1.772 -2.375 34.336 1.00 41.23 N
HETATM 2 C1 QTT A 602 2.186 -1.989 35.672 1.00 38.26 C
HETATM 3 O1 QTT A 602 5.279 -5.886 36.039 1.00 25.03 O
...
HETATM 29 C02 QTT A 602 -2.261 -5.769 33.449 1.00 54.51 C
TER 30 QTT A 602
END
可以通过与PDB文件中的对应部分比对,确认一致性。
转换为SDF格式
你可以使用OpenBabel将提取的PDB文件转换为SDF文件。如果尚未安装OpenBabel,可以通过以下命令安装:
conda install -c conda-forge openbabel
然后使用以下命令将PDB文件转换为SDF:
obabel ligand_1_QTT_602_A.pdb -O 7A0C_QTT_602_A.sdf
obabel ligand_2_QTT_601_B.pdb -O 7A0C_QTT_601_B.sdf
这样便可以生成相应的SDF文件。
1.3 SDF与MOL2之间的相互转化
使用 OpenBabel 可以非常方便地在SDF和MOL2文件之间进行转换。以下是两种转换的示例:
1.将SDF文件转换为MOL2文件
obabel input.sdf -O output.mol2
input.sdf:SDF格式的输入文件。-O output.mol2:指定生成的MOL2文件名。
2.将MOL2文件转换为SDF文件
obabel -i mol2 input.mol2 -o sdf -O output.sdf
input.mol2:MOL2格式的输入文件。-O output.sdf:指定生成的SDF文件名。
2 从分子文件生成SMILES表示
在本节中,我们将探讨如何通过不同的分子文件格式(如 SDF、PDB)生成分子的 SMILES 表示。我们将分别使用 RDKit 和 OpenBabel 两种工具,展示如何通过代码实现这一过程,并在最后加入分子的可视化环节。
提示:以下代码建议在 Jupyter Notebook 中运行,以便于查看输出和可视化结果。
2.1 使用RDKit从分子文件生成SMILES
RDKit 是一个功能强大的开源化学信息学工具包,能够处理多种分子文件格式并生成SMILES表示。
1.从SDF文件生成SMILES
首先,我们通过RDKit从 SDF 文件中读取分子并生成SMILES表示。
from rdkit import Chem
# 读取SDF文件
supplier = Chem.SDMolSupplier('7A0C_QTT_602_A.sdf')
# 遍历每个分子,提取其SMILES表示
for mol in supplier:
if mol is not None:
smiles = Chem.MolToSmiles(mol)
print("SMILES表示:", smiles)
SMILES表示: Cc1cccc(CN(C)[C@H]2CCCC[C@@H]2N(CC(=O)O)Cc2cccc(C)n2)n1
2.从Mol2文件生成SMILES(不推荐)
接下来,我们从 Mol2 文件中读取分子并生成SMILES。
from rdkit import Chem
# 读取Mol2文件
mol_supplier = Chem.MolFromMol2File('7A0C_QTT_602_A.mol2')
# 检查是否读取成功
if mol_supplier is not None:
smiles = Chem.MolToSmiles(mol_supplier)
print(smiles)
SMILES表示: [C]c1[c][c][c]c([C]N([C])[C]2[C][C][C][C][C]2N([C]C(=O)[O-])[C]c2[c][c][c]c([C])n2)n1
在该输出中,我们可以看到大量的 [C] 这样的占位符。这是因为 Mol2 文件格式可以存储详细的化学信息,包括原子的部分电荷、键的类型、立体化学信息等。然而,使用 Chem.MolFromMol2File 函数读取 Mol2 文件时,RDKit 可能无法正确解析其中的立体化学信息。这导致生成的 SMILES 表示不完全一致,尤其是某些关键的立体化学和原子信息被简化或丢失。因此,RDKit 输出的 SMILES 中出现了大量 [C] 占位符,这并不代表正确的分子结构。
为了生成更准确的 SMILES 表示,推荐使用更适合的文件格式(如 SDF 文件),或者借助其他化学信息处理工具(如 OpenBabel)对 Mol2 文件进行转换和处理。
3.从PDB文件生成SMILES
RDKit 也支持从 PDB 文件中生成SMILES表示。
from rdkit import Chem
# 从PDB文件读取分子,`removeHs=False` 参数确保在读取PDB文件时保留氢原子,这有助于更准确地重建分子结构。
mol = Chem.MolFromPDBFile('ligand_1_QTT_602_A.pdb', removeHs=False)
# 检查是否成功读取,并生成SMILES
if mol is not None:
smiles = Chem.MolToSmiles(mol)
print("SMILES表示:", smiles)
else:
print("无法读取PDB文件。")
SMILES表示: CC1CCCC(CN(C)[C@H]2CCCC[C@@H]2N(CC(O)O)CC2CCCC(C)N2)N1
2.2 使用OpenBabel从分子文件生成SMILES
1.从SDF文件生成SMILES:
obabel 7A0C_QTT_602_A.sdf -o smi -O output.smi
输出的 output.smi 文件内容示例:
N(C)(Cc1cccc(n1)C)[C@H]1CCCC[C@@H]1N(CC(=O)O)Cc1cccc(n1)C ligand_1_QTT_602_A.pdb
2.从PDB文件生成SMILES:
obabel ligand_1_QTT_602_A.pdb -o smi -O output.smi
输出的 output.smi 文件内容示例:
N(C)(Cc1cccc(n1)C)[C@H]1CCCC[C@@H]1N(CC(=O)O)Cc1cccc(n1)C ligand_1_QTT_602_A.pdb
2.3 常见的文件格式与 SMILES 生成的关系
| 文件格式 | 主要用途 | 优点 | 缺点 | 常用于生成 SMILES 吗? |
|---|---|---|---|---|
| SDF | 化学结构、药物设计 | 保存详细的键连接、手性、氢原子信息 | 文件较大,处理大规模化学数据库时效率较低 | 是 |
| PDB | 生物大分子、配体相互作用 | 详细的三维坐标 | 小分子的键连接信息较弱,手性信息不如SDF详细 | 是 |
| Mol2 | 分子对接、药物设计 | 保存部分电荷、分子类型 | 可能丢失手性或氢原子信息,键连接可能不够详细 | 不常用 |
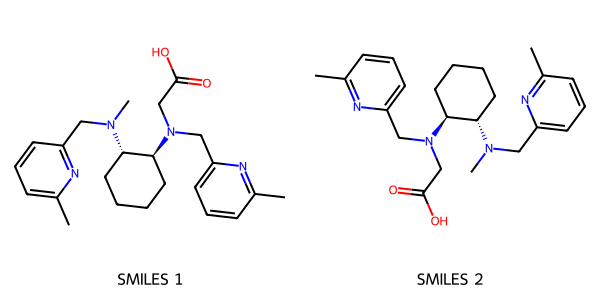
2.4 SMILES的可视化
生成SMILES后,我们可以使用 RDKit 对分子进行可视化,帮助我们直观地理解分子结构。
使用RDKit绘制分子结构
from rdkit import Chem
from rdkit.Chem import Draw
# SMILES 字符串
smiles1 = 'Cc1cccc(CN(C)[C@H]2CCCC[C@@H]2N(CC(=O)O)Cc2cccc(C)n2)n1' # RDKit sdf
smiles2 = 'N(C)(Cc1cccc(n1)C)[C@H]1CCCC[C@@H]1N(CC(=O)O)Cc1cccc(n1)C' # OpenBabel pdb
# 从 SMILES 生成分子对象
mol1 = Chem.MolFromSmiles(smiles1)
mol2 = Chem.MolFromSmiles(smiles2)
# 将分子对象放入列表
mols = [mol1, mol2]
# 添加标题,方便辨识
legends = ['SMILES 1', 'SMILES 2']
# 绘制分子并显示
img = Draw.MolsToGridImage(mols, molsPerRow=2, subImgSize=(300, 300), legends=legends)
from IPython.display import display
display(img)
在Jupyter Notebook中,直接运行上述代码会显示分子的二维结构图。
规范化 SMILES
从上面的SMILES以及分子对象图可以看出,同一个结构的SMILES可能有多种,为了判断不同的 SMILES 是否表示相同的分子,可以使用 RDKit 的规范化 SMILES 功能:
# 生成规范化的 SMILES
canonical_smiles1 = Chem.MolToSmiles(mol1, canonical=True)
canonical_smiles2 = Chem.MolToSmiles(mol2, canonical=True)
print('规范化 SMILES 1:', canonical_smiles1)
print('规范化 SMILES 2:', canonical_smiles2)
# 比较规范化 SMILES 是否相同
if canonical_smiles1 == canonical_smiles2:
print('两个 SMILES 表示相同的分子。')
else:
print('两个 SMILES 表示不同的分子。')
输出:
规范化 SMILES 1: Cc1cccc(CN(C)[C@H]2CCCC[C@@H]2N(CC(=O)O)Cc2cccc(C)n2)n1
规范化 SMILES 2: Cc1cccc(CN(C)[C@H]2CCCC[C@@H]2N(CC(=O)O)Cc2cccc(C)n2)n1
两个 SMILES 表示相同的分子。
显示分子带有原子编号的结构
有时,我们希望在分子图中显示原子编号,方便对照分析。
from rdkit.Chem.Draw import IPythonConsole # Enables RDKit drawing in Jupyter
from rdkit.Chem import rdDepictor
from rdkit.Chem.Draw import rdMolDraw2D
# 为分子生成2D坐标
rdDepictor.Compute2DCoords(mol1)
# 初始化绘图对象
drawer = rdMolDraw2D.MolDraw2DCairo(500, 500)
opts = drawer.drawOptions()
opts.addAtomIndices = True # 显示原子编号
# 绘制分子
drawer.DrawMolecule(mol1)
drawer.FinishDrawing()
# 显示图像
from IPython.display import Image
Image(drawer.GetDrawingText())

总结
本文通过实例展示了如何从PDB文件中提取配体分子,并利用Biopython、OpenBabel等工具将这些分子保存为SDF或Mol2文件。我们深入探讨了使用RDKit和OpenBabel生成SMILES表示的方法,并分析了不同分子文件格式的优缺点。RDKit在处理SDF文件时能够准确生成SMILES表示,但对Mol2文件的解析可能存在局限性。我们还展示了如何使用RDKit进行分子结构的可视化,并引导读者通过规范化SMILES判断不同分子是否相同。


















 668
668

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











