







胡说八道还发网上, 我有罪
所有组分统一使用GAFF力场, 对离子支持的并没有那么好, 我不会联用GAFF和Merz/KBFF力场. 离子液体, 高浓度多种离子(尤其少见的像稀土离子)的溶液.容易被质疑.
问题
1.需要模拟什么体系, 体系中各种组分的摩尔比或者浓度?
2.想要通过模拟说明什么问题?
3.给两篇该领域实验加分子动力学模拟的文献以供参考.
模拟流程规范
1.力场文件构建, 工作目录sobtop
准备好分子的.mol2文件放入sobtop文件夹, 执行bash RESP_noopt.sh 分子.mol 电荷 自旋多重度, 得到.chg文件. 执行sobtop, 生成拓扑GAFF, 1 2 4 回车 回车. 将.itp文件中的字段添加ffnonbonded.itp中.
2.溶液盒子的构建, 工作目录pdb文件数据库
准备优化好的pdb文件, 加入pdb结构文件夹, 编写文件夹中的in.inp文件. 修改gmx_test脚本路径, 执行gmx_test脚本得到进行eq模拟的结构.
3.正式模拟, 工作目录/public/home/likai/gmx/工作名/
将sobtop目录的拓扑文件链接到这些文件夹内, 将grompp.mdp, toppol.放入文件夹进行长时间模拟.
基础知识
宏观的性质基本都有一套分子动力学的模拟方法, 如溶解性, 介电常数. 模拟的时间尺度<2000 ns. 每次1fs,模拟时间尺度普遍在几十ns, 通常需要模拟上千万步.
gromacs的问题, 不支持复杂力场MMFF94, EAM势, ReaxFF, 对可极化力场支持不充分.
Centos7以后的版本装不了2020年以后的版本, 对gcc要求变高了, 22版之后有CP2K接口.
高温应该用更小的步长, 带氢的1ps, 约束H可以用2ps. 没有H步长可以设置更大.
简单力场势能表达式
简单位力场势能表达式:
E
total
=
∑
bonds
1
2
k
b
(
r
−
r
0
)
2
+
∑
angles
1
2
k
a
(
θ
−
θ
0
)
2
+
∑
dihedrals
V
2
[
1
+
cos
(
n
φ
−
φ
0
)
]
+
∑
improper
1
2
k
i
(
ξ
−
ξ
0
)
2
+
∑
ADB
[
C
12
r
A
B
−
C
6
r
A
B
6
]
+
∑
A
>
B
q
A
q
B
4
π
ε
0
r
A
B
E^{\text{total} } = \sum_{\text{bonds} } \frac{1}{2}k^b (r - r_0)^2 + \sum_{\text{angles} } \frac{1}{2}k^a (\theta - \theta_0)^2 + \sum_{\text{dihedrals} } \frac{V}{2} \left[ 1 + \cos (n\varphi - \varphi_0) \right] + \sum_{\text{improper} } \frac{1}{2}k^i (\xi - \xi_0)^2 + \sum_{\text{ADB} } \left[ \frac{C_{12} }{r_{AB} } - \frac{C_6}{r_{AB}^6} \right] + \sum_{A > B} \frac{q_A q_B}{4 \pi \varepsilon_0 r_{AB} }
Etotal=bonds∑21kb(r−r0)2+angles∑21ka(θ−θ0)2+dihedrals∑2V[1+cos(nφ−φ0)]+improper∑21ki(ξ−ξ0)2+ADB∑[rABC12−rAB6C6]+A>B∑4πε0rABqAqB
成键项:
bonds, angles:成键相和角度相, 常用谐振式表示.
dibedrals:二面角相, 旋转360复原, 所以用周期函数拟合得到.
improper:1-4键相互作用
非键项:占据了总计算量的90%
范德华相互作用:最常用L-J势, 第一项为交换互斥, 第二项为色散作用, 量化证明精确的六次方关系, 第一项使用9-10次精度更高, 但是12次方是后面的平方, 节省了计算量.
库伦相互作用:只有对范德华表面外静电势重现性好的原子电荷才能准确描述分子间静电相互作用.
能量最小化积分算法:最好的是Velocity Verlet, 但是gromacs用的是leap-frog算法. 步长应小于振动频率最高的周期的1/10, 与H相关的键10fs, 所以用1fs. 约束氢可以2fs.
控温:Velocity-rescale最好, 传统Nose-Hoover模拟前期出现震荡且遍历性差.
压浴:Stochastic Cell Rescaling最好, 比较新, 用的人不多. 对液体τ=0.5即可. PR压浴类似Nose-Hoover有震荡问题.
水模型:SPC/E三点水模型, OPC3是最好的三点水模型, OPC是最好的四点水模型(都不是可极化水模型).
力场文件:GAFF力场与AMBER兼容, 描述各种有机小分子好, 描述蛋白质核酸不如AMBER. 描述电解液体系(带小分子, 阴阳离子是稳妥的选择).
模拟示例:模拟电解质对氢键网络的影响
体系:KCF3SO3的浓度为 5 mol/L, 六聚乙二醇(PEG)的浓度为 10 mol/L.
结构文件来源:PEG(1.在pubchem上检索2.在molview上搜索修改, 可以进行初步优化). 发现在pubchem上下载的乙醇结构原子在同一平面上导致高斯无法优化. H2O是gview建的(用力场对应的水结构应该更准确).
建模:使用packmol构建溶剂盒子得到conf.pdb文件(packmol得到的几乎是充分混合的). packmol潜在问题是建模不能考虑电荷, 当加入多种阴离子怀疑可能会导致距离过近出现模拟崩溃. in.inp文件示例如下, 注意单位是埃, 区别与.gro文件的nm.
tolerance 2.0 # 容差, 两个分子之间的最小距离为2.0埃
filetype pdb # pdb无法添加盒子信息, 也许用.xyz会更好(没试过)
output conf.pdb
# add_box_sides 1.2盒子尺寸增加额外的边界宽度, 但是不填充, 避免分子在边界上的重叠或受限.
structure ce.pdb
number 6
inside cube 0.0 0.0 0.0 48.8
end structure
structure h.pdb
number 165
inside cube 0.0 0.0 0.0 48.8
end structure
structure so4.pdb
number 90
inside cube 0.0 0.0 0.0 48.8
end structure
structure po4.pdb
number 3
inside cube 0.0 0.0 0.0 48.8
end structure
structure h2o.pdb
number 3336
inside cube 0.0 0.0 0.0 48.8
end structure ```
用win的packmol有时出现不提示也没输出就一直卡在那. 不知道是packmol的问题还是电脑的问题. 盒子设置的太小需要更多计算时间, 如果一直运行不成功那就是盒子设置的太小了. 盒子的大小对计算影响很大, 太大的盒子导致npt模拟时, 盒子体积缩小十分缓慢. 太小会导致运行之初就报错, 一运行就终止很可能是初始结构或者参数不合理.
sobtop构建拓扑文件:需要得到原子上的电荷以计算静电相互作用, 使用RESP2电荷计算得到.chg文件. 将.mol2和.chg按照顺序载入, 可以得到基于RESP2电荷的GAFF力场文件. mutiwfn要改默认设置以调用gaussian和formcheck. 具体操作:
使用Multiwfn的examples目录下的RESP2_noopt.sh文件, 执行
bash RESP2_noopt.sh CF3SO3.mol2 -1 1 #-1, 1分别对应于电荷和自旋多重度
bash RESP2_noopt.sh CF3SO3.mol2 -1 1 #-1, 1分别对应于电荷和自旋多重度
模拟过程
先做能量极小化防止初始结构太不合理导致模拟崩溃. 平衡相跑完根据密度, 温度…判断稳定, 注意压力变化剧烈, 模拟常压相差十几bar都算正常, 将最后一帧作为初始跑产生相. 产生相就是时间更长的平衡相.

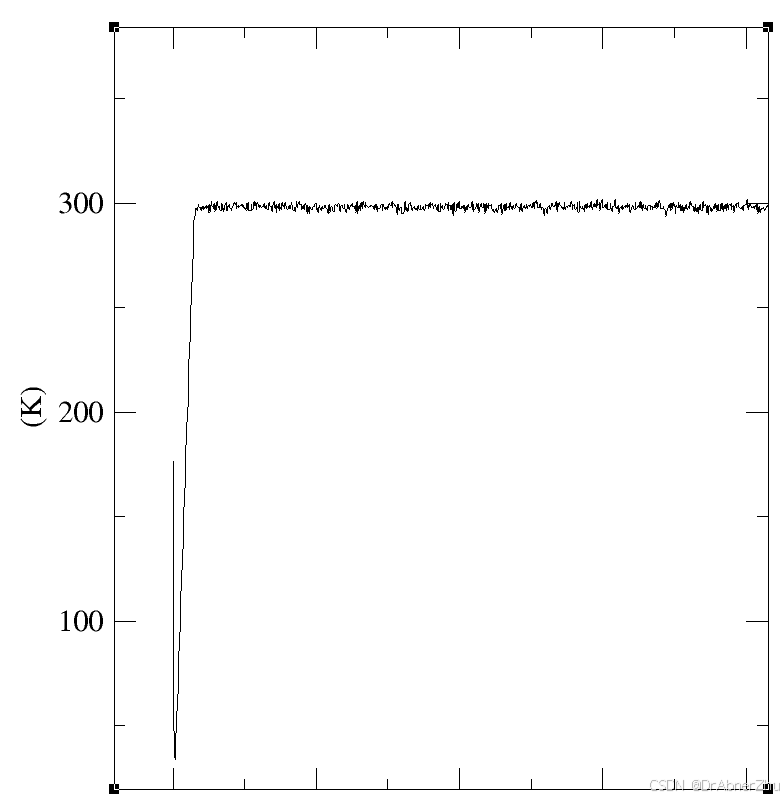
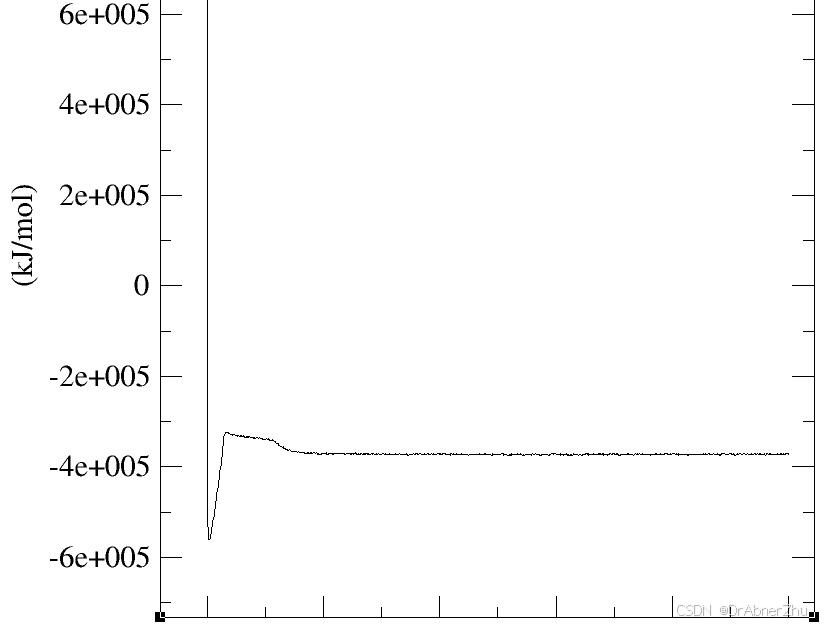
修改默认颜色, 将gview_color.tcl文件放入VMD目录下, 在VMD的vmd.rc文件末尾加入一行proc gview {} {source gview_color.tcl} , 命令行输入gview, 在representations的color method使用Element.以使用gview配色, Zn的颜色似乎没加载成功, 更改颜色, 创建名为name ZN的, color method, 选择colorID, 颜色为iceblue和gview颜色一致.
关掉坐标轴标签, 背景换成白色color Display Background white
轨迹文件会出现乱成键的情况, 用gmx转化一下gmx trjconv -s md.tpr -f md.xtc -o md_fixed.xtc -pbc mol或者执行mol bondsrecalc all, topo retypebonds, 只对当前帧生效.
添加盒子(这个貌似只能终端执行)pbc box -color black -width 2
键长键角:分析某些键长键角在Graphic labels可以作图, save导出文本格式作图.
加载到ovito,执行gmx trjconv -s topol.tpr -f traj_comp.xtc -o traj.pdb, -f traj_comp.xtc要加, 因为默认找的值traj.xtc(也可能是traj.tr
但是xtc的默认文件名是traj_comp, 感觉是有点小bug)-o traj.pdb要加, 否则输出.xtc文件.
氢键数
gmx hbond -f traj_comp.xtc -s topol.tpr -nomerge
输出
SS1 1.593777e+04 9.083148e+01 1.229362e+00 3.241 23.643
SS1是平均每一帧形成的氢键数量, 计算方法ss12/分子数
*不加-nomerge,一个氢键给体只能形成一个氢键
1.593777e+042/10200=3.125, 纯水正常数值为3.6,添加剂减少了水分子氢键的数量,抑制了质子的传递
qtgrace hbnum.xvg进行作图,gmx analyze -f hbnum.xvg可以对.xvg进行分析,输出与上面一样
水的MSD
注win版做msd会出现selection is parsesd但是不输出的情况, ctrl+d什么的也无效. 这时候可以用echo "6" | gmx msd -f .\traj_comp.xtc -s .\topol.tpr -o msd.xvg注意不加-o也不输出.
输出
D[SOL] = 0.8798 (+/- 0.0052) (1e-5 cm^2/s)
TCL脚本分析
resname指定残基名, atom指定原子名, 做个能量最小化, 把confout的名称给VMD, 最好是改拓扑文件的残基名.
为了可视化某种原子周围显示的效果, same residue as (within 5 of name ZN) 为原子名为ZN周围5埃以内的整个残基创建一类. 切换为正视图截图, display resetview
VMD处理VASP的XDATCAR
MS建模, 得到CIF文件, CIF文件中的H2O是按分子排序的, 用vesta转POSCAR会导致元素合并在一起, 用openbabel不会出现这个问题, 得到POSCAR和.gro或者.pdb文件.(这两文件作为第一帧提供残基名和原子名). 对.gro/.pdb文件的残基名和原子类型进行编辑. 不过这样VASP的POSCAR原子就很长
结构文件与包含的信息(emm, 不知道自己在说啥了)
pdb文件可以包含盒子信息, CRYST1 81.27 81.30 81.29 90.00 90.00 90.00 P 1 1它不包含成键信息, 适用于packmol. mol2文件不包含盒子信息, 包含成键信息, 适用于mutiwfn和sobtop, 但是既然不包含盒子信息, 转换得到的.gro文件也不包含盒子信息.
就是REMARK
CRYST1 81.27 81.30 81.29 90.00 90.00 90.00 P 1 1
90.00 P 1 1它不包含成键信息, 适用于packmol. mol2文件不包含盒子信息, 包含成键信息, 适用于mutiwfn和sobtop, 但是既然不包含盒子信息, 转换得到的.gro文件也不包含盒子信息.
就是REMARK
CRYST1 81.27 81.30 81.29 90.00 90.00 90.00 P 1 1
离子液体模拟, 力场也要非常讲究, 现有的非极化离子液体力场没有能够非常完美地描述其结构的, 水分子也有很多模型, 而且并不是任何一种都能准确描述水的结构. 两者结合到一起能够正确描述离子液体和水的相互作用就更难了. 你得首先单独模拟纯的离子液体和水, 把它们的结构和性质弄对, 然后再模拟混合物. 可能需要反复尝试各种力场和水分子模型. 有可能需要考虑力场极化和分子间电子转移.

















 610
610

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









