






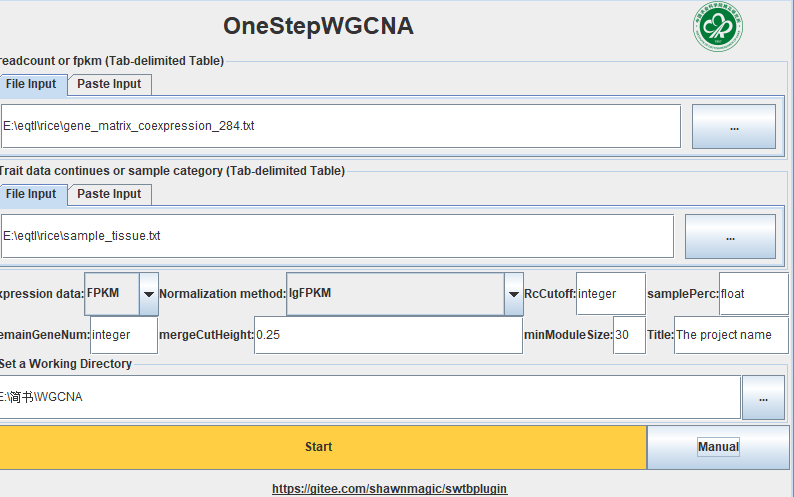
今天发现TBtools上有了个神器 Rserver 。这意味我们可以把别人写好的R脚本直接拿过来运行。于是用OneStepWGCNA这个插件试验一下。本文主要记录试验过程和可能遇到的一些问题,主要参考『TBtools-plugin』OneStepWGCNA插件这篇文章。想要了解WGCNA的具体含义和流程可以参考:
WGCNA分析,简单全面的最新教程
一文学会WGCNA分析
WGCNA官方网站WGCNA算法研究笔记
OneStepWGCNA 的准备文件和详细参数
虽然题目有点标题党,但这个插件真的方便,真的感谢这个插件的作者。
工具准备
TBtools
Rserver: 需要在TBtools的用户群里下载,一共181M,相当于一个独立的R环境,具体安装方法参考Rserver 插件 for TBtools。用户群可以在公众号:生信药丸里找到。
OneStepWGCNA插件:可以在TBtools的plugin store下载到。建议先下载plugin store里的plugin store at high speed。因为在plugin store里下载插件太慢了,太慢了,太慢了。
#数据准备
OneStepWGCNA 的输入文件是基因表达矩阵和表型数据(或样品分类数据)。这里用了RED数据库里的284个日本晴的不同部位,不同处理的表达矩阵(FPKM数据)。样品分类数据用每个样品取的组织。
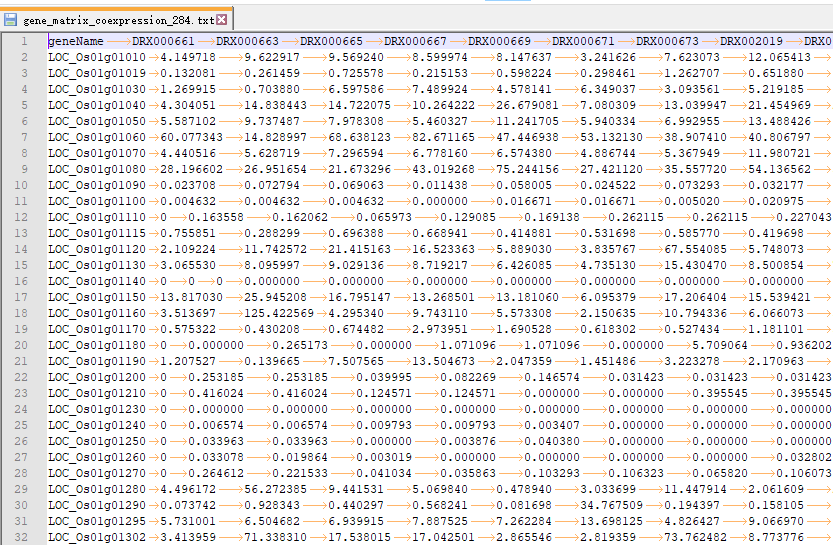
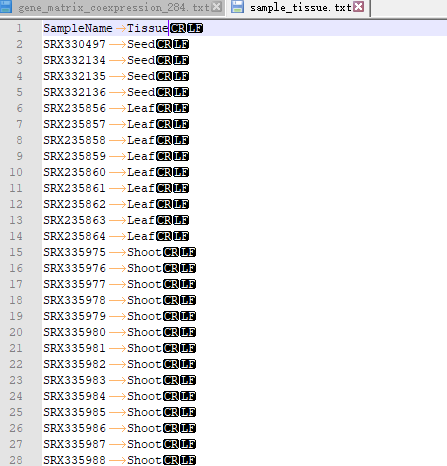
第一次跑的时候遇到的坑

第一次包发现GenomeInfoDb这个包没下下来,R文件里也没有申明需要这个包。我在TBtools的插件目录下OneStepWGCNA的R脚本里加上了:
if (!require(‘GenomeInfoDb’)) install.packages(‘GenomeInfoDb’)
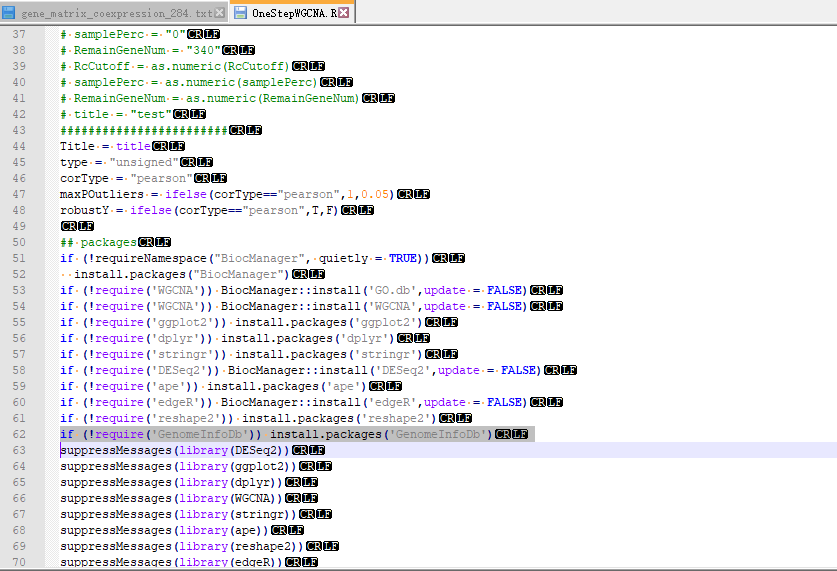
再跑一次就成功了
这个TBtools插件文件夹位置一般在C:\Users\names\ .TBtools.Plugin\OneStepWGCNA
OneStepWGCNA 参数详解
readcount or fpkm 是表达矩阵,以Tab为分割符,行是样本,列是基因。
Trait data 是表型文件或分类文件,可以有多个表型,可以是连续性也可以是离散。如果要把多分类性状和连续性状都混到一起,可以把多分类性状转为one-hot编码,这个用下excel就能完成。
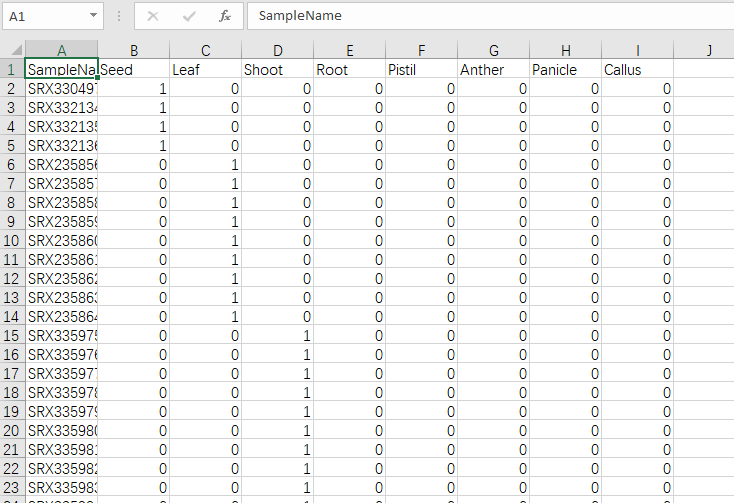
Expression data 是表达矩阵数据类型,count数据是不能直接用来做WGCNA的
Normalization method 是表达矩阵转换的方式,如果是count数据可以选varianceStabilizingTransformation或log10(CPM+1),如果是FPKM可以选rawFPKM,不经过转换,和取对数。建议都试试,看看哪个效果更好。
RcCutoff 是用来除噪声的,我看原作者的教程里大概意思是count推荐10,FPKM推荐0.2,当然,这个参数还是要随着测序深度和具体生物学问题而改变的。
samplePerc 和上面的参数配对的,在0-1之间,如果是0.5,RcCutoff 是0.2 代表想要 50%的样品的FPKM>0.2。
RemainGeneNum 是保留多少基因进行WGCNA,写0或小于0就是啥都没了,会报错。
mergeCutHeight 是合并模块的阈值, 0.25的意思就是把相关系数大于0.75的模块合并。
minModuleSize 是指最少一个模块要包含30个基因。
输出结果
Sample_clustering 样品聚类结果

这里如果出现明显的离群样本,记住样本的名字,在表达矩阵和trait矩阵中删掉对应样本再跑一次
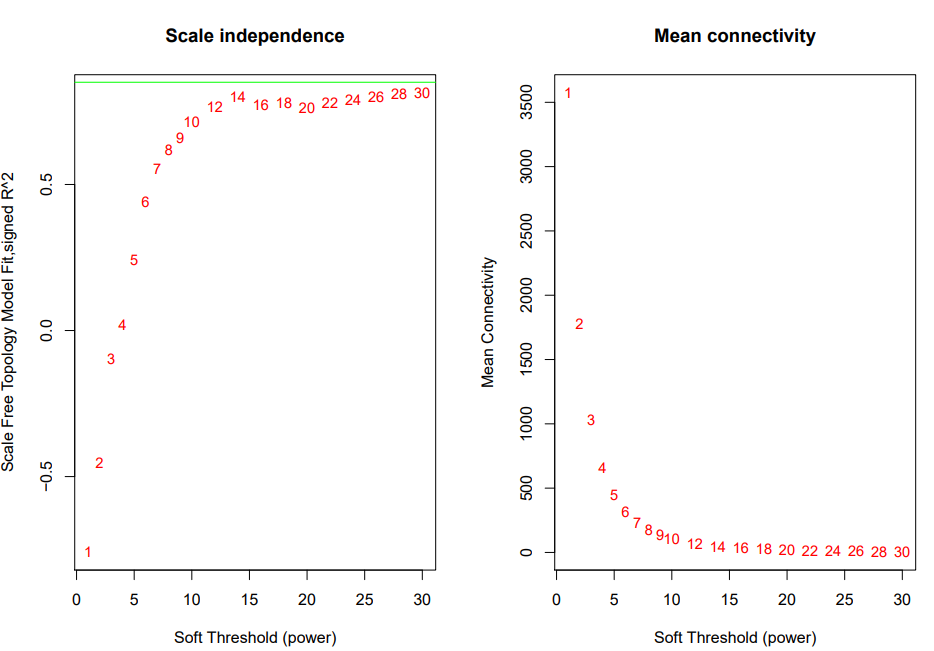
绿线为0.8,这里没有筛的很好的阈值,就选了经验阈值14来做power进行后续计算。
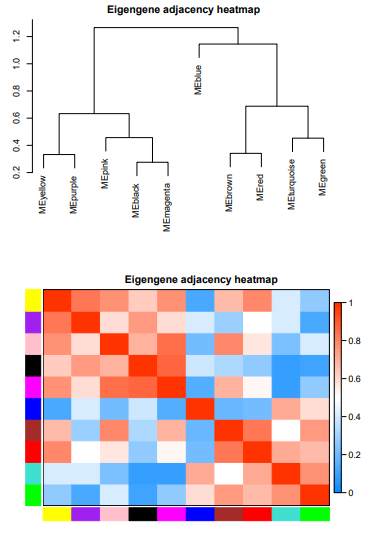
一共找到了10个模块,该结果主要反应模块间的相关性。
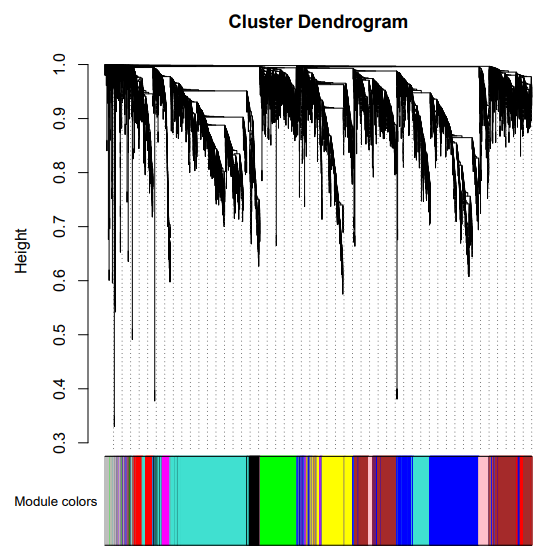
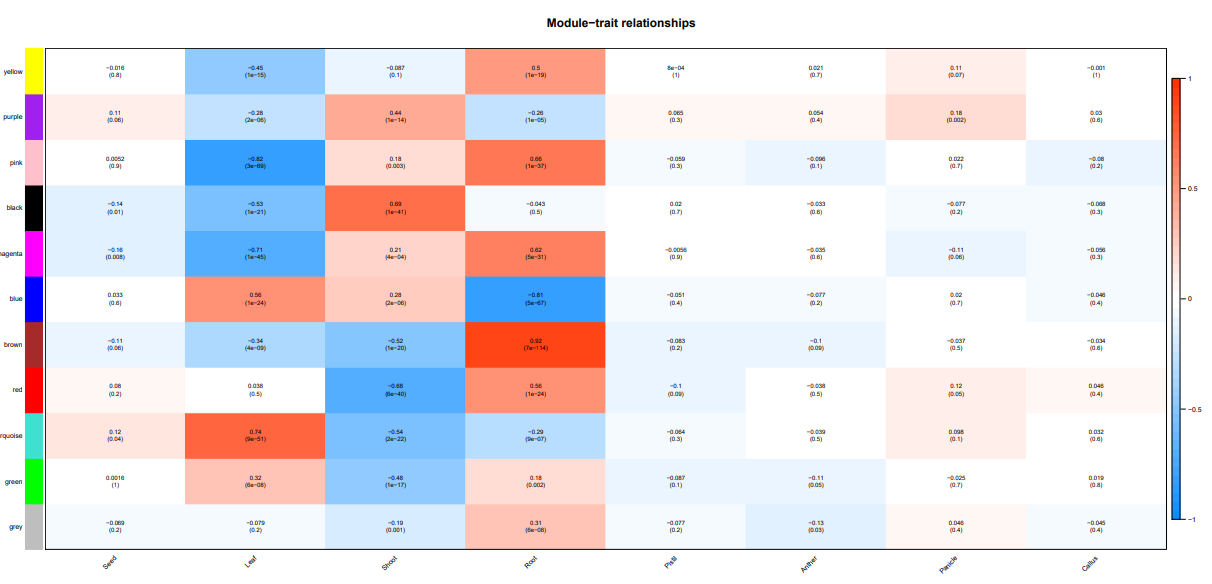
发现棕色模块和根的相关性最高,其他种子,花药,穗的部位都没有找到相关性高的模块。一个可能是由于这几个部位的样本量少,另一方面本身不同处理对表达量的影响也很大。这里只是试一试,并不能反应真实的科学结论。
可以拿表型关联上的模块做后续的分析,如GO,KEGG啥的,巧的是富集分析也能用TBtools做。
最后再唠叨两句
我发现最后结果里还有很多信息没有被存下来,我想看选几个基因画TOM图,或者画个网络图,或者是图的字体太小了,看不清想重新画,该咋办?
最粗暴的办法还是把整个网络保存下来,想要啥,自己整。
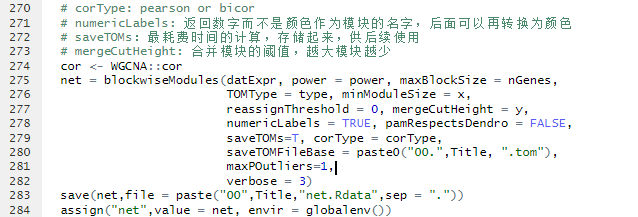
以下代码主要来源于 WGCNA分析,简单全面的最新教程
library(WGCNA)
#把存入的Rdata都读进来,总能用上的 \xk one_traits是我设的Title name,要改
load("00.one_traits.datatraitbase.Rdata")
load("00.one_traits.net.Rdata")
load("00.one_traits.datatraitbase.Rdata")
load("00one_traits.Modular.Rdata")
load("one_traits.tom-block.1.RData")
Title<-"one_traits"
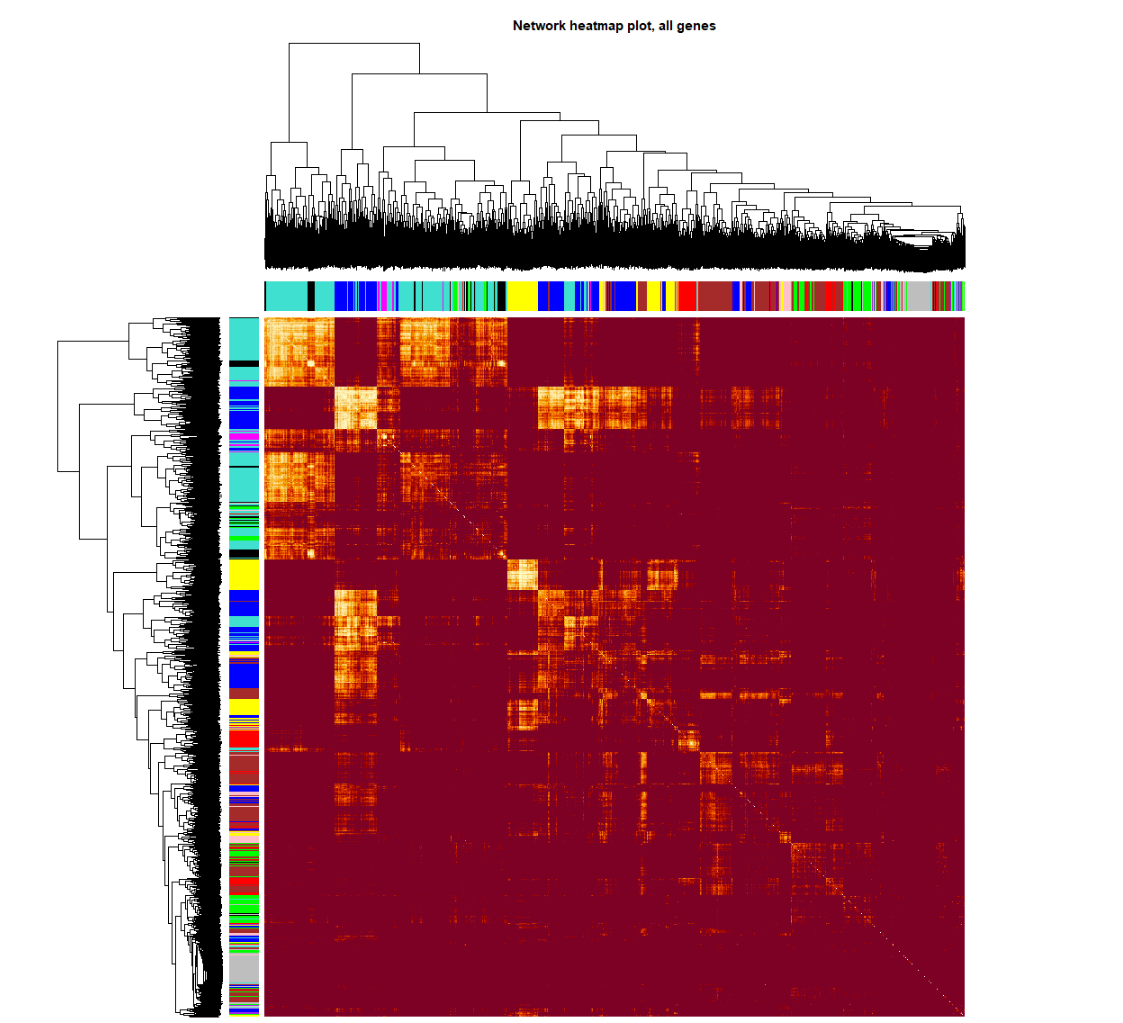
###TOM图
moduleColors = labels2colors(net$colors)
TOM <- as.matrix(TOM)
dissTOM = 1-TOM
# Transform dissTOM with a power to make moderately strong
# connections more visible in the heatmap
plotTOM = dissTOM^7
# Set diagonal to NA for a nicer plot
diag(plotTOM) = NA
# Call the plot function
# 这一部分特别耗时,行列同时做层级聚类
TOMplot(plotTOM, net$dendrograms, moduleColors,
main = "Network heatmap plot, all genes")
###导出Cytoscape 图格式
probes = colnames(datExpr)
dimnames(TOM) <- list(probes, probes)
# Export the network into edge and node list files Cytoscape can read
# threshold 默认为0.5, 可以根据自己的需要调整,也可以都导出后在
# cytoscape中再调整
cyt = exportNetworkToCytoscape(TOM,
edgeFile = paste(Title, ".edges.txt", sep=""),
nodeFile = paste(Title, ".nodes.txt", sep=""),
weighted = TRUE, threshold = 0.5,
nodeNames = probes, nodeAttr = moduleColors)
## 模块内基因与表型数据关联
#关联样品性状的二元变量时,设置
robustY = ifelse(corType=="pearson",T,F)
if (corType=="pearsoon") {
geneModuleMembership = as.data.frame(cor(datExpr, MEs_col, use = "p"))
MMPvalue = as.data.frame(corPvalueStudent(
as.matrix(geneModuleMembership), nSamples))
} else {
geneModuleMembershipA = bicorAndPvalue(datExpr, MEs_col, robustY=robustY)
geneModuleMembership = geneModuleMembershipA$bicor
MMPvalue = geneModuleMembershipA$p
}
# 计算性状与基因的相关性矩阵
## 只有连续型性状才能进行计算,如果是离散变量,在构建样品表时就转为0-1矩阵。
traitData<-read.table(traitsfile,header=T,sep='\t') #traitsfile 表型文件
if (corType=="pearsoon") {
geneTraitCor = as.data.frame(cor(datExpr, traitData, use = "p"))
geneTraitP = as.data.frame(corPvalueStudent(
as.matrix(geneTraitCor), nSamples))
} else {
geneTraitCorA = bicorAndPvalue(datExpr, traitData, robustY=robustY)
geneTraitCor = as.data.frame(geneTraitCorA$bicor)
geneTraitP = as.data.frame(geneTraitCorA$p)
}
## Warning in bicor(x, y, use = use, ...): bicor: zero MAD in variable 'y'.
## Pearson correlation was used for individual columns with zero (or missing)
## MAD.
# 最后把两个相关性矩阵联合起来,指定感兴趣模块进行分析
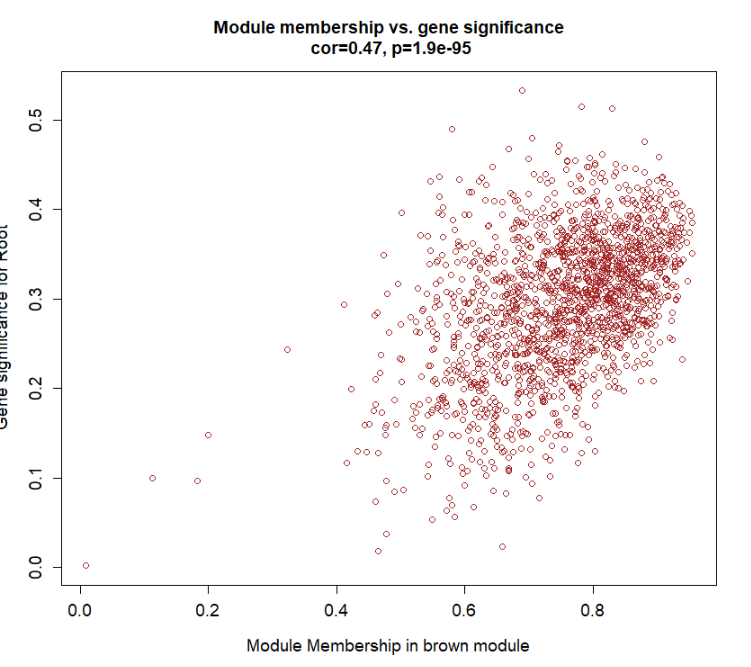
module = "brown"#感兴趣的模块
pheno = "Root" #感兴趣的性状
modNames = substring(colnames(MEs_col), 3)
# 获取关注的列
module_column = match(module, modNames)
pheno_column = match(pheno,colnames(traitData))
# 获取模块内的基因
moduleGenes = moduleColors == module
# 与性状高度相关的基因,也是与性状相关的模型的关键基因
verboseScatterplot(abs(geneModuleMembership[moduleGenes, module_column]),
abs(geneTraitCor[moduleGenes, pheno_column]),
xlab = paste("Module Membership in", module, "module"),
ylab = paste("Gene significance for", pheno),
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)

















 3479
3479

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









