






1写在前面
最近实在是忙的不行,根本没时间更新,一到家就只想睡觉。🥹
今天写个最近用到的分析方法,Weighted correlation network analysis (WGCNA),是非常经典的生信分析方法了,现在被引有9913次了,马上就要破万啦。😘
网上相关的教程也是不胜枚举,但多多少少是有些不尽人意的地方,有的少步骤,有的代码不全。😅
这里在仔细阅读了官方手册后,在这里和大家一起认真地step by step研究一下,查缺补漏吧。🥰
2用到的包
rm(list = ls())
library(tidyverse)
library(WGCNA)
3示例数据
数据是雌性小鼠肝脏的基因表达谱,来自这篇paper:👇
Ghazalpour A, Doss S, Zhang B, et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2(8):e130. doi:10.1371/journal.pgen.0020130
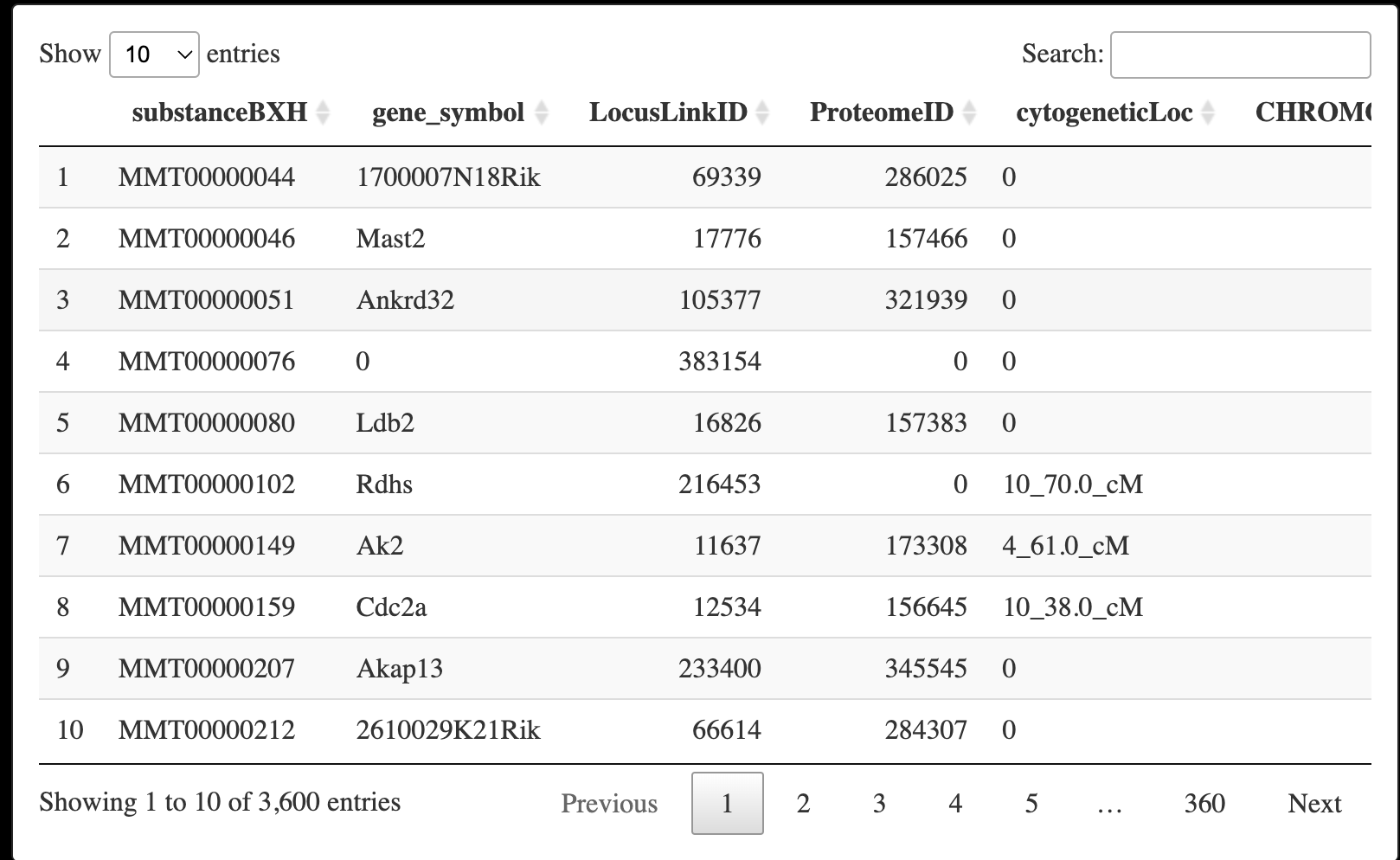
dat <- read.csv("./FemaleLiver-Data/LiverFemale3600.csv")
DT::datatable(dat)
4整理数据
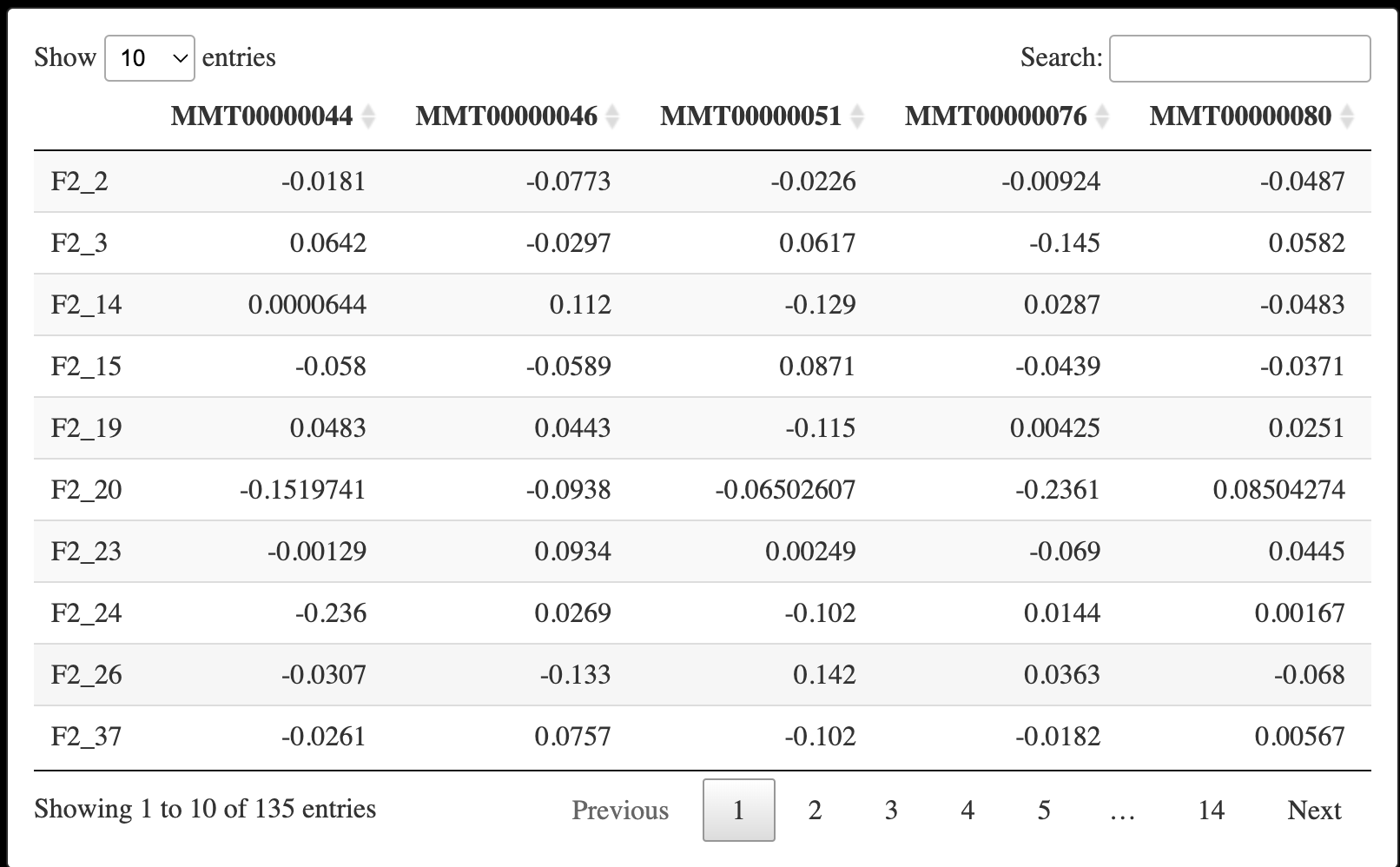
我们先提取表达矩阵,这里是需要转置的。😅
datExpr0 <- as.data.frame(t(dat[, -c(1:8)]))
names(datExpr0) <- dat$substanceBXH
rownames(datExpr0) <- names(dat)[-c(1:8)]
DT::datatable(datExpr0)
5基因或样本过滤
有一些表达值过低的基因或样本,我们是需要过滤掉的,包里也是提供了相应的函数,我们看一下吧。😂
5.1 查看是否有不好的基因或样本
我们的数据里没有不好的基因或者样本。😘
gsg <- goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK

5.2 自动化过滤
这里提供一个if语句,显示不好的基因或者样本,进行自动化过滤。🥳
if (!gsg$allOK)
{
## 打印已删除的基因和样本名称
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}
6样本聚类
接着我们需要对样本进行聚类,有一些outlier的样本可能还需要去除掉。🤨
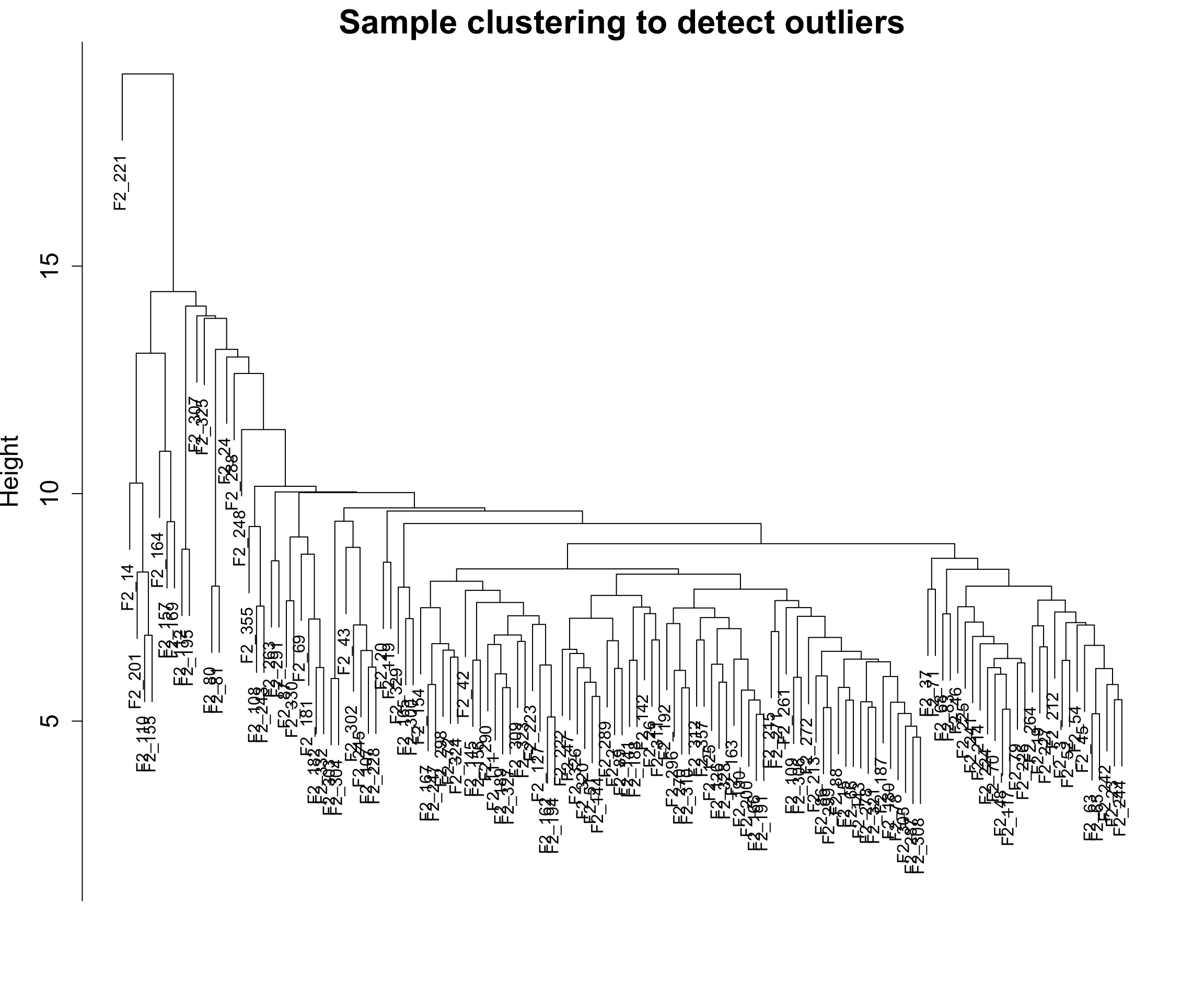
6.1 绘制聚类树
聚类的方法很多,这里整理一下:👇
ward.D", "ward.D2", "single", "complete", "average", "mcquitty", "median", "centroid"
sampleTree <- hclust(dist(datExpr0), method = "average");
plot(sampleTree,
main = "Sample clustering to detect outliers",
sub="",
xlab="",
cex.lab = 1.5, cex.axis = 1.5, cex.main = 2)
6.2 画个红线
这里我们有一个聚类比较差的样本,我们把它去掉吧。🥹
plo9888888=t(sampleTree,
main = "Sample clustering to detect outliers",
sub="",
xlab="",
cex.lab = 1.5, cex.axis = 1.5, cex.main = 2
)
abline(h = 15, col = "red")
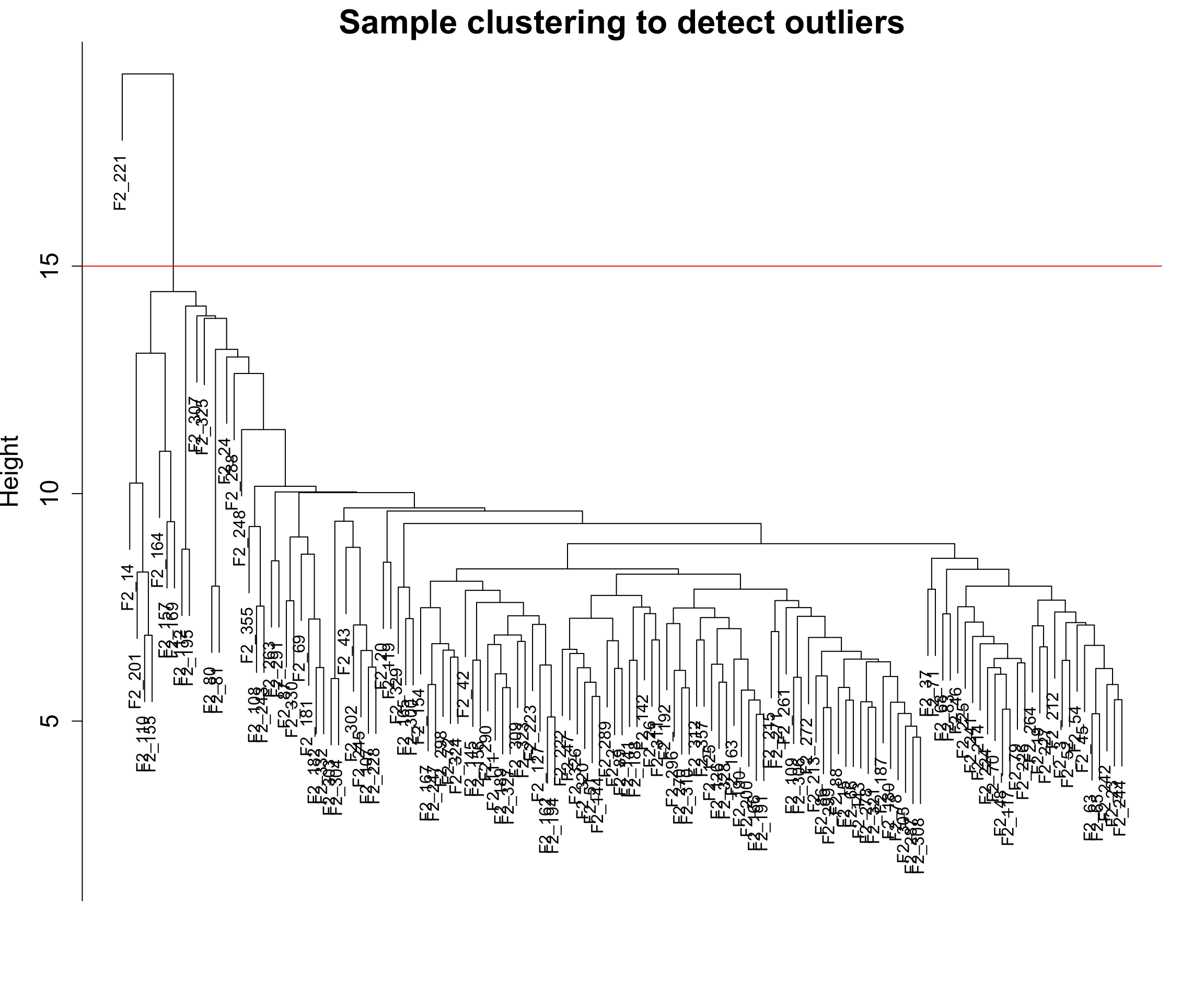
6.3 去除聚类异常的样本
clust <- cutreeStatic(sampleTree, cutHeight = 15, minSize = 10)
table(clust)

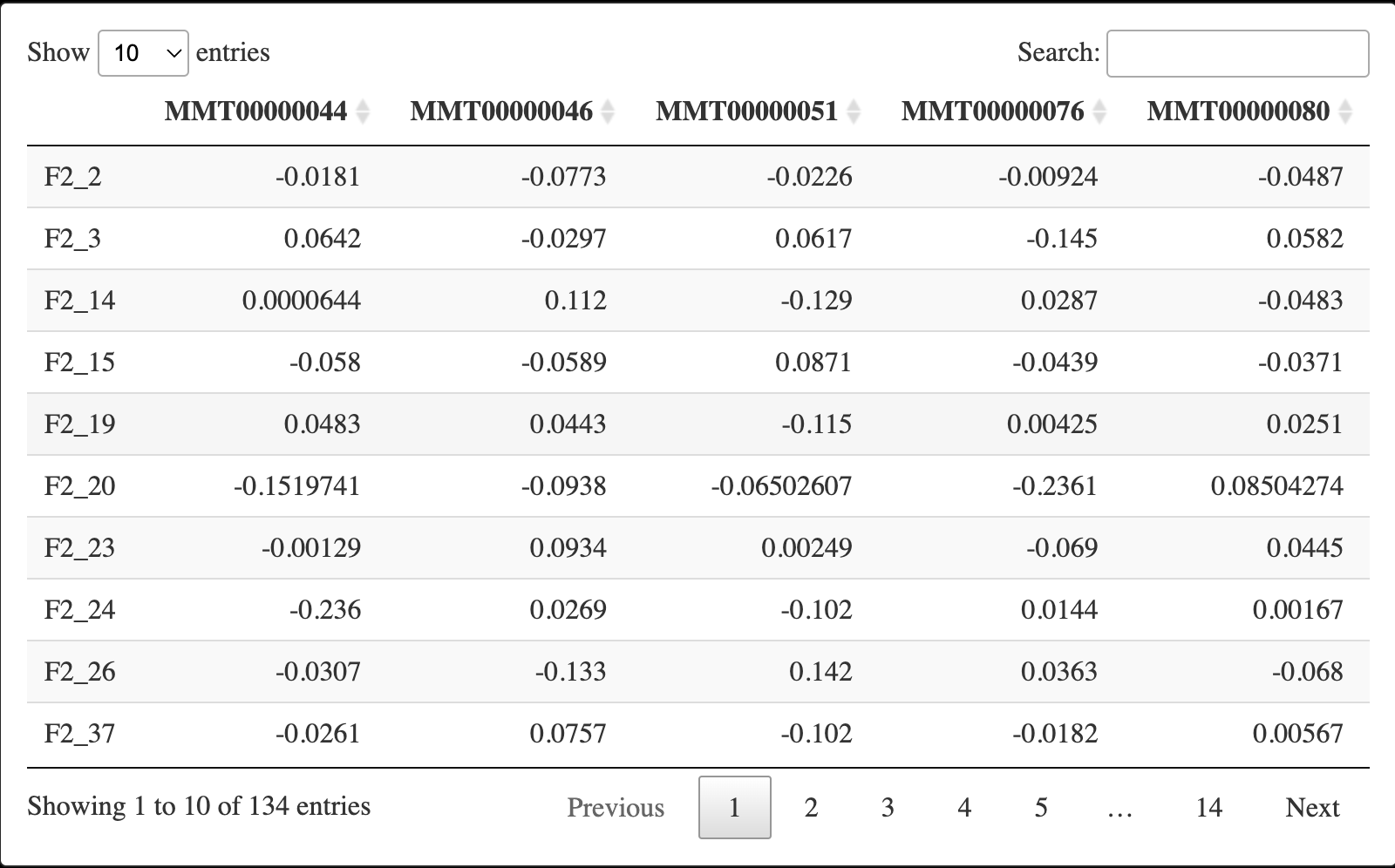
6.4 提取过滤后矩阵
keepSamples <- (clust == 1)
datExpr <- datExpr0[keepSamples, ]
nGenes <- ncol(datExpr)
nSamples <- nrow(datExpr)
DT::datatable(datExpr)
7加载临床/性状数据
接着我们把临床或性状数据(traits)导入进来,和前面的聚类树一起绘图。🥳
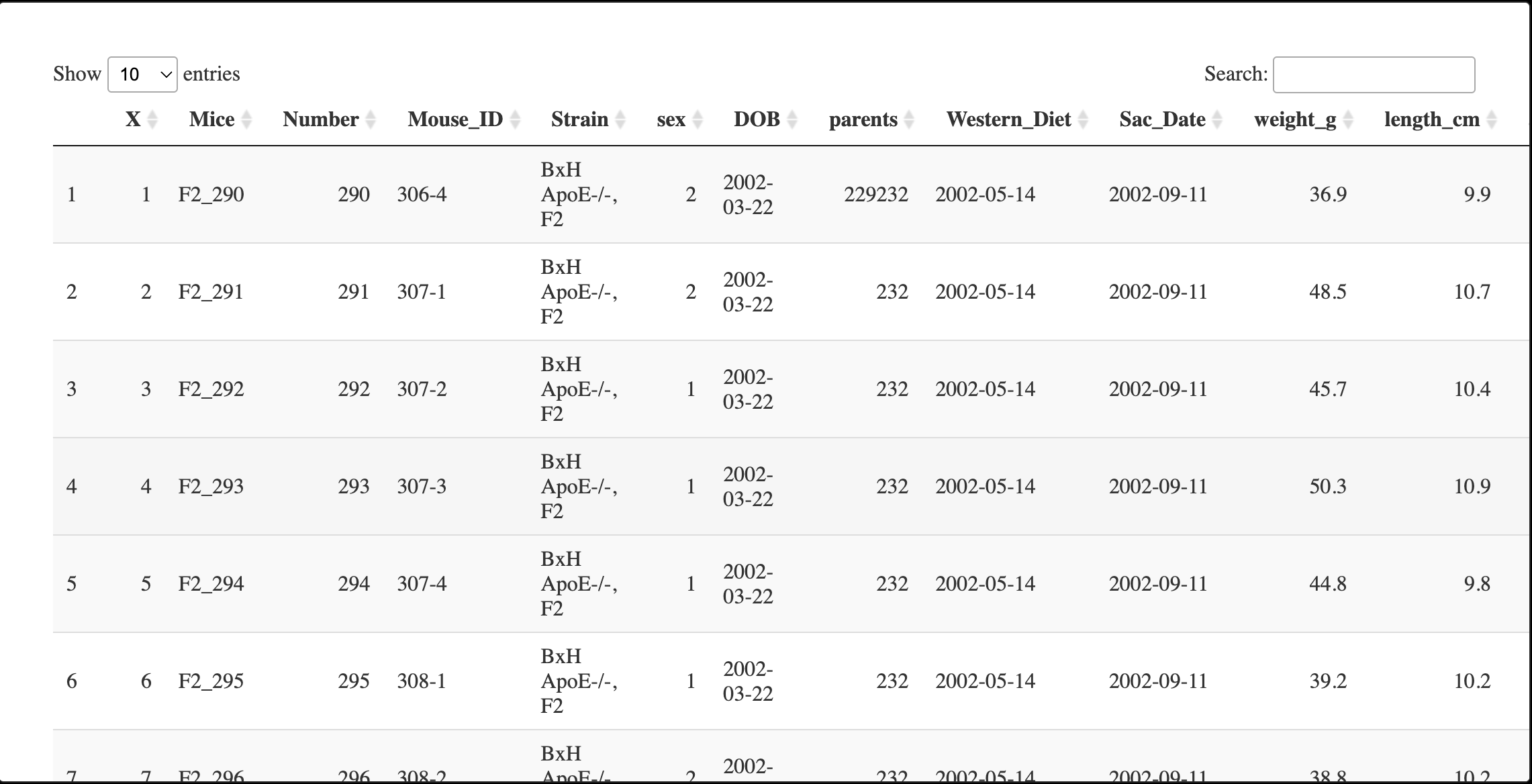
7.1 读入traits
traitData <- read.csv("./FemaleLiver-Data/ClinicalTraits.csv");
DT::datatable(traitData)
7.2 整理traits
我们把一些不需要的traits去掉,只保留我们自己需要的,这里需要和样本名一一对应上。🤪
allTraits <- traitData[, -c(31, 16)]
allTraits <- allTraits[, c(2, 11:36) ]
femaleSamples <- rownames(datExpr)
traitRows <- match(femaleSamples, allTraits$Mice)
datTraits <- allTraits[traitRows, -1]
rownames(datTraits) <- allTraits[traitRows, 1]
collectGarbage()
DT::datatable(datTraits)
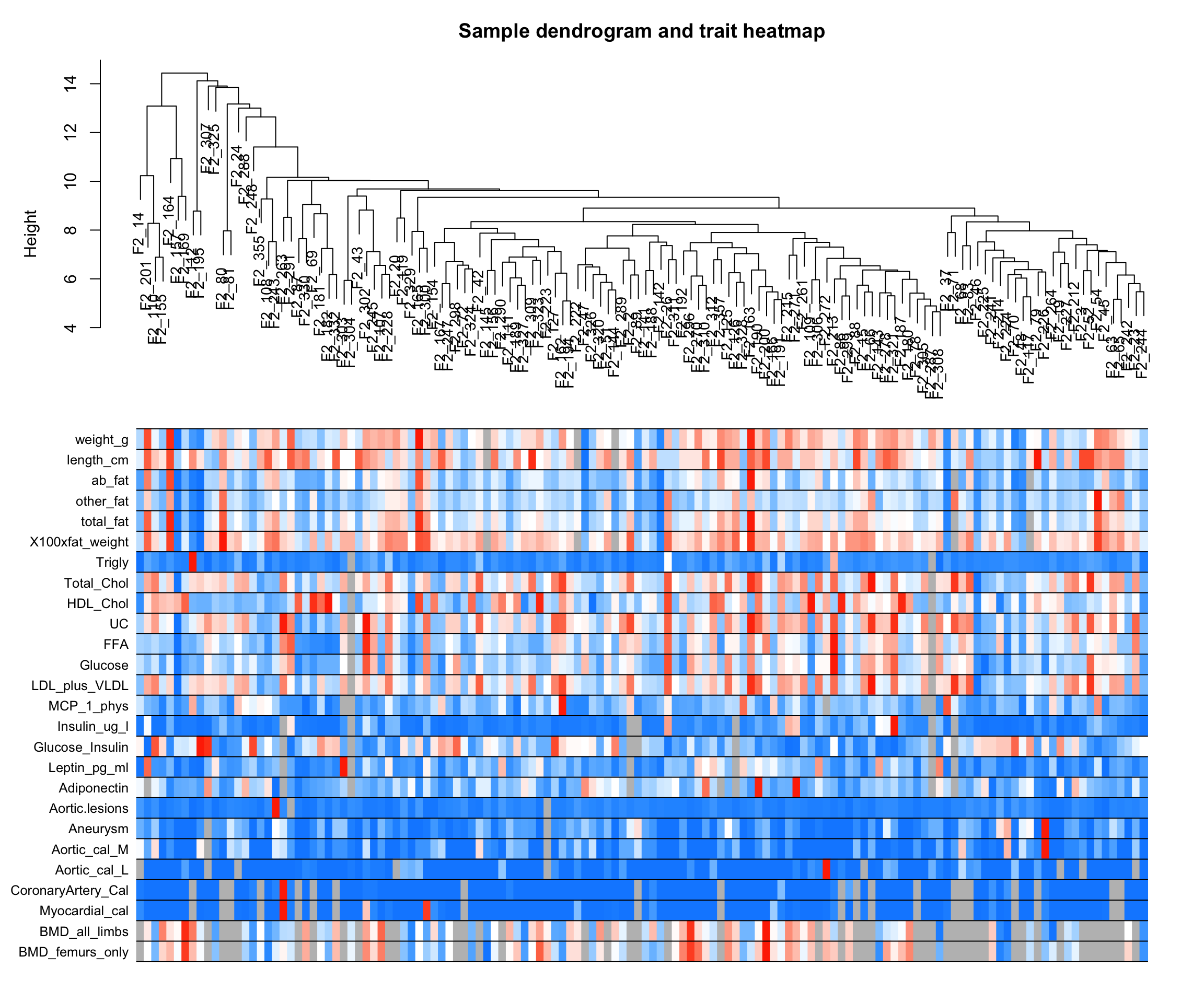
8绘制最终聚类树
sampleTree2 <- hclust(dist(datExpr), method = "average")
traitColors <- numbers2colors(datTraits,
signed = F,
colors = greenWhiteRed(100)
)
plotDendroAndColors(sampleTree2,
traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")
9save一下
这里我们保存一下数据,下期继续。😘
save(datExpr, datTraits, file = "FemaleLiver-01-dataInput.RData")
10补充一下
现在很多paper都是先做差异基因分析,然后将DEGs提取出来做WGCNA,其实这种方法原作者并不推荐,还是推荐大家将所有基因初步过滤后进行WGCNA的分析,原文如下:👇
"We do not recommend filtering genes by differential expression. WGCNA is designed to be an unsupervised analysis method that clusters genes based on their expression profiles. Filtering genes by differential expression will lead to a set of correlated genes that will essentially form a single (or a few highly correlated) modules."
11如何引用
📍
Langfelder, P., Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). https://doi.org/10.1186/1471-2105-9-559

点个在看吧各位~ ✐.ɴɪᴄᴇ ᴅᴀʏ 〰
📍 🤩 ComplexHeatmap | 颜狗写的高颜值热图代码!
📍 🤥 ComplexHeatmap | 你的热图注释还挤在一起看不清吗!?
📍 🤨 Google | 谷歌翻译崩了我们怎么办!?(附完美解决方案)
📍 🤩 scRNA-seq | 吐血整理的单细胞入门教程
📍 🤣 NetworkD3 | 让我们一起画个动态的桑基图吧~
📍 🤩 RColorBrewer | 再多的配色也能轻松搞定!~
📍 🧐 rms | 批量完成你的线性回归
📍 🤩 CMplot | 完美复刻Nature上的曼哈顿图
📍 🤠 Network | 高颜值动态网络可视化工具
📍 🤗 boxjitter | 完美复刻Nature上的高颜值统计图
📍 🤫 linkET | 完美解决ggcor安装失败方案(附教程)
📍 ......
本文由 mdnice 多平台发布

















 5044
5044

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









