







如果你使用 GENE-IS: Saira Afzal et al。 ,2016请引用这篇研究文章。GENE-IS: time-efficient and accurate analysis of viral integration events in large-scale gene therapy data. Molecular Therapy - Nucleic Acids 2016, vol. 6:133-139. DOI:https://doi.org/10.1016/j.omtn.2016.12.001
GENE-IS 是从临床和临床前基因治疗研究的下一代测序数据中提取整合位点的管道。它是专门为了接受来自不同方案如 LAM (线性扩增介导) PCR 和靶向测序(SureSelect/AGILENT)方法的测序读数而设计的。
我该怎么办?
Installation
获取和运行GENE-IS最简单的方法是克隆目前的存储库
mkdir path_to_location
cd path_to_location
git clone https://github.com/G100DKFZ/gene-is.git
cd gene-is
Testing
为了测试安装是否成功,我们准备了 testGenis.sh,这是一个简单的脚本,可以对一组减少的数据集运行快速分析。
在终端上键入以下命令,将目录更改为脚本
cd /path_to_location/gene-is/scripts
# export the location of gene-is
export GENIS=/path_to_location/gene-is
# Run test suite by following command
./testGenis.sh
终端上会出现这些选项;
1) Targeted Sequencing Pair BWA 4) All
2) Targeted Sequencing Single 5) Clear
3) LAM-PCR 6) Quit
在终端1运行目标测序配对终端模式类型的测试并按回车键。如果安装成功,以下信息将出现在终端“Targeted Sequencing Pair worked as expected!目标测序对工作正常!”
要在终端2运行目标测序单端模式类型的测试并按回车键。如果安装成功,以下信息将出现在终端“Targeted Sequencing Single end worked as expected!目标测序单端工作正常!”
在终端3运行 LAM-PCR 配对终端模式类型测试并按回车键。如果安装成功,下面的消息将出现在终端“ LAM-PCR Pair worked as expected!LAM-PCR 对工作正常!”
为了测试用于 GENE-IS 基准测试的 Manuscript 中使用的数据集,请参见“/path _ to _ location/GENE-IS/testFiles”目录中的“ README”文件
但是我目前的结果出现了以下报错

解决办法,这是因为.sh结尾的脚本文件需要用bash运行,
bash testGenis.sh
成功出现以下结果:
1) Targeted Sequencing Pair BWA
2) Targeted Sequencing Single
3) LAM-PCR
4) All
5) Clear
6) Quit
依赖
第三方工具
GENE-IS 依赖于几个第三方工具,这些工具是开源的,可以免费使用。所有这些工具都已经在 $GENIS/tools/bin 目录的 GENE-IS 包中提供。此文件夹被称为配置文件中第三方工具的默认位置
#############################################################################
## Third-party tools
#Provide path to these third-party tools
#############################################################################
#Provide path to the BWA aligner
aligner = $GENIS/tools/bin/bwa
#Path to the secondary aligner. (BLAT)
blatAligner = $GENIS/tools/bin/blat
#Path to the trimming and filtering tool (Skewer)
skewer = $GENIS/tools/bin/skewer
#Path to the Samtools
samtools= $GENIS/tools/bin/samtools
#Path to the bedtools
bedTools= $GENIS/tools/bin/bedtools
对于用户信息,这里提供了工具名称和相关链接; 工具版本 URL
BWA 0.7.4 http://sourceforge.net/projects/bio-bwa/files/?source=navbar
Bedtools 2.17.0 https://code.google.com/p/bedtools/downloads/detail?name=BEDTools.v2.17.0.tar.gz&can=2&q=
Samtools 0.1.19 http://samtools.sourceforge.net/
BLAT v.35 http://users.soe.ucsc.edu/~kent/src/blatSrc35.zip
Skewer 0.1.117 http://sourceforge.net/projects/skewer/files/Binaries/
这里我选择了2,运行单端数据的运算,出现以下结果:
......................................................
Tue 19 Mar 2024 06:25:20 PM CST
###################################################################
Pre-proceesing (Quality Filtering and Adapter Trimming) in progress...
perl /home/mdisk/****/00.software/path_to_location/gene-is/scripts/filteringTrimming.pl -f /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/testData.TS.pair1.fastq.gz -qual 20 -adaptF GATCGGAAGAGCACACGTCTGAACTCCAGTCAC -sOut filtTrim -o /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -sk /home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/skewer
Quality value is 20.
Results will be stored in /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/
/home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/skewer -x GATCGGAAGAGCACACGTCTGAACTCCAGTCAC -q 20 -l 50 -o /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/testData.TS.pair1.fastq.gz
Parameters used:
-- 3' end adapter sequence (-x): GATCGGAAGAGCACACGTCTGAACTCCAGTCAC
-- maximum error ratio allowed (-r): 0.100
-- maximum indel error ratio allowed (-d): 0.030
-- end quality threshold (-q): 20
-- minimum read length allowed after trimming (-l): 20
-- file format (-f): Sanger/Illumina 1.8+ FASTQ (auto detected)
-- minimum overlap length for adapter detection (-k): 3
Tue Mar 19 18:25:20 2024 >> started
|> | (0.66%)
Tue Mar 19 18:25:21 2024 >> done (0.541s)
50000 reads processed; of these:
4541 ( 9.08%) short reads filtered out after trimming by size control
6447 (12.89%) empty reads filtered out after trimming by size control
39012 (78.02%) reads available; of these:
33385 (85.58%) trimmed reads available after processing
5627 (14.42%) untrimmed reads available after processing
log has been saved to "/home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.log".
###################################################################
Alignment in process...
=======>>>>>>>>>>>> 0
perl -I /home/mdisk/****/00.software/path_to_location/gene-is/lib /home/mdisk/****/00.software/path_to_location/gene-is/scripts/alignment.pl -p 8 -f filtTrim.fastq -gv /home/mdisk/****/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa -a /home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/bwa -aOut completAlignment -o /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -t AGILENT -sam /home/mdisk/*****/00.software/path_to_location/gene-is/tools/bin/samtools
Alignemnt type AGILENT
BWA is used as Aligner
/home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/bwa mem -M -t 8 /home/mdisk/****/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq > /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment.sam
[M::main_mem] read 39012 sequences (6961052 bp)...
[main] Version: 0.7.4-r385
[main] CMD: /home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/bwa mem -M -t 8 /home/mdisk/****/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq
[main] Real time: 0.902 sec; CPU: 5.069 sec
/home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/samtools: error while loading shared libraries: libncurses.so.5: cannot open shared object file: No such file or directory
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/samtools: error while loading shared libraries: libncurses.so.5: cannot open shared object file: No such file or directory
###################################################################
IS extraction and post-processing in progress...
perl -I /home/mdisk/*/00.software/path_to_location/gene-is/lib /home/mdisk/*/00.software/path_to_location/gene-is/scripts/extractIS.pl -aIn completAlignment.sam -o /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -s /home/mdisk/*/00.software/path_to_location/gene-is/scripts -v VECTOR -vecFile /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/VECTOR.fa -genFile /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa -fOut filtTrim.fastq -aBWA /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa -t AGILENT -i /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa.2bit -bla /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/blat -minIden 95 -range 10
Results will be stored in /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/
sed -i /@SQb/d /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment.sam
awk '/home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa ~ /S/' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS.sam
sed -i '/^$/d' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS.sam
CountLines=8981
echo 8981 > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt
python /home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_MS_Correction_Oct2014.py /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected.sam
File "/home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_MS_Correction_Oct2014.py", line 19
print i
^^^^^^^
SyntaxError: Missing parentheses in call to 'print'. Did you mean print(...)?
sort -u -k1,1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted.sam
cut -f 1,2,3,4,5,6,7,8,9,10,11,12 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1.sam
awk '{ sub(/0$/, +, /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/) }1' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1.sam | awk '{ sub(/256$/, +, /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/) }1' | awk '{ sub(/16$/, -, /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/) }1' | awk '{ sub(/272$/, -, /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/) }1' | awk '{ sub(/0$/, +, filtTrim.fastq) }1' | awk '{ sub(/256$/, +, filtTrim.fastq) }1' | awk '{ sub(/16$/, -, filtTrim.fastq) }1' | awk '{ sub(/272$/, -, filtTrim.fastq) }1' > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1_strand.sam
awk -v OFS=t 'completAlignment.sam=completAlignment.sam' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1_strand.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1_strand1.sam
python /home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_SeprateSM_Oct2014.py /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1_strand1.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM.sam
File "/home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_SeprateSM_Oct2014.py", line 21
print i
^^^^^^^
SyntaxError: Missing parentheses in call to 'print'. Did you mean print(...)?
sed -i '/^$/d' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM.sam
python /home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_SeprateMS_Oct2014.py /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_corrected_sorted1_strand1.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS.sam
File "/home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_SeprateMS_Oct2014.py", line 21
print i
^^^^^^^
SyntaxError: Missing parentheses in call to 'print'. Did you mean print(...)?
sed -i '/^$/d' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS.sam
python /home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_SM_Sextraction.py /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted.sam
File "/home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_SM_Sextraction.py", line 23
span=cig1[:S1]
TabError: inconsistent use of tabs and spaces in indentation
python /home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_MS_Sextraction.py /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted.sam
File "/home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_MS_Sextraction.py", line 23
span=cig1[M1+1:S1]
TabError: inconsistent use of tabs and spaces in indentation
awk -v OFS=t 'completAlignment.sam=completAlignment.sam' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1.sam
awk -v OFS=t 'completAlignment.sam=completAlignment.sam' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1.sam
awk '(completAlignment.sam3 >= 20 )' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1aa.sam
awk '(completAlignment.sam3 >= 20 )' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1aa.sam
cut -f1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1aa.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact-ids.txt
awk -vExact=/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact-ids.txt 'BEGIN{while((getline<Exact)>0)l[@completAlignment.sam]=1}NR%2==1{f=l[completAlignment.sam]?1:0}f' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact.fastq
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/VECTOR.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam
[main] Version: 0.7.4-r385
[main] CMD: /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/VECTOR.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact.fastq
[main] Real time: 0.009 sec; CPU: 0.002 sec
sort /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam | uniq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly1.sam
cut -f1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly1.sam | sort | uniq -u > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam.ids
awk -vExactVecOnly=/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam.ids 'BEGIN{while((getline<ExactVecOnly)>0)l[@completAlignment.sam]=1}NR%2==1{f=l[completAlignment.sam]?1:0}f' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact1.fastq
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact1.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam
[main] Version: 0.7.4-r385
[main] CMD: /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact1.fastq
[main] Real time: 0.109 sec; CPU: 0.004 sec
sort /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam | uniq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly1.sam
cut -f1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly1.sam | sort | uniq -u > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam.ids
awk 'NR==FNR{tgts[completAlignment.sam]; next} completAlignment.sam in tgts' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam.ids /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1aa.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1a.sam
cut -f1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1aa.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact-ids.txt
awk -vExact=/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact-ids.txt 'BEGIN{while((getline<Exact)>0)l[@completAlignment.sam]=1}NR%4==1{f=l[completAlignment.sam]?1:0}f' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact.fastq
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/VECTOR.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam
[main] Version: 0.7.4-r385
[main] CMD: /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/VECTOR.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact.fastq
[main] Real time: 0.006 sec; CPU: 0.002 sec
sort /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam | uniq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly1.sam
cut -f1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly1.sam | sort | uniq -u > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam.ids
awk -vExactVecOnly=/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactVecOnly.sam.ids 'BEGIN{while((getline<ExactVecOnly)>0)l[@completAlignment.sam]=1}NR%4==1{f=l[completAlignment.sam]?1:0}f' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact1.fastq
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact1.fastq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam
[main] Version: 0.7.4-r385
[main] CMD: /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa mem -M /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Exact1.fastq
[main] Real time: 0.007 sec; CPU: 0.003 sec
sort /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam | uniq > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly1.sam
cut -f1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly1.sam | sort | uniq -u > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam.ids
awk 'NR==FNR{tgts[completAlignment.sam]; next} completAlignment.sam in tgts' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ExactGenOnly.sam.ids /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1aa.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1a.sam
python /home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_MS_MPosCorrectionOct2014.py /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//Lines.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1a.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1a_correctedMpos0.sam
File "/home/mdisk/*/00.software/path_to_location/gene-is/scripts/CIGAR_MS_MPosCorrectionOct2014.py", line 24
spanM1=cig1[:M1]
TabError: inconsistent use of tabs and spaces in indentation
sed -e s/ /t/g /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1a_correctedMpos0.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1a_correctedMpos.sam
awk -Ft 'BEGIN { OFS = t } {completAlignment.sam6=1; print}' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_MS_Sextracted1a_correctedMpos.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//MS_MS.sam
awk -Ft 'BEGIN { OFS = t } {completAlignment.sam5=0; print}' /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1a.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_SM.sam
cut -f 15 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_SM.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_SM.txt
paste /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_idChrIS.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_strand.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_Sspan.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_read.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_SM.txt | sed 's/\t/@/g' > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_header.txt
cut -f 14 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_onlyS_SM_Sextracted1a.sam > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_Sseq.txt
paste /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_header.txt /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//SM_Sseq.txt | sed -e 's/^/>/' | sed 's/\t/\n/g' > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S_SM.fa
cat /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S_MS.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S_SM.fa > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S.fa
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/blat /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa.2bit /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S.fa -out=blast8 -minIdentity=95 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S.bst
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/blat: error while loading shared libraries: libpng12.so.0: cannot open shared object file: No such file or directory
echo VECTOR
VECTOR
awk -Ft -v vectorStr=VECTOR -f /home/mdisk/*/00.software/path_to_location/gene-is/scripts/extractIS.awk /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S.bst | grep VECTOR > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDup.csv
awk: /home/mdisk/*/00.software/path_to_location/gene-is/scripts/extractIS.awk:64: fatal: cannot open file `/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S.bst' for reading (No such file or directory)
mv: cannot stat '/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment_S.bst': No such file or directory
sort: cannot read: /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtered.bst: No such file or directory
sort -k2,2 -k3,3 -k4,4 -k5,5 -k6,6 -k7,7 -k8,8 -k9,9 -k10,10 -u /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDup.csv > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDupSingle0.csv
sort -k1,1 -u /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDupSingle0.csv > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDupSingle.csv
cut -d -f 1,2,3,4,5,6,7 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDupSingle.csv | awk -v vectorName=VECTOR -f /home/mdisk/*/00.software/path_to_location/gene-is/scripts/formatIS.awk | sort -k4nr > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDup.csv.total
sort -k1,1 -k2,2n /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDup.csv.total | awk -v range=10 -f /home/mdisk/*/00.software/path_to_location/gene-is/scripts/solveIS.awk > /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDup.csv.total.results
bash /home/mdisk/*/00.software/path_to_location/gene-is/scripts/extractSingleEndIS.sh completAlignment.sam /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ /home/mdisk/*/00.software/path_to_location/gene-is/scripts VECTOR /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/VECTOR.fa /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testOnlyGenome.fa filtTrim.fastq /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/blat /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bwa /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa.2bit 95 10
No extra filtering...
###################################################################
Multiple aligned reads processing TS ...
bash /home/mdisk/*/00.software/path_to_location/gene-is/scripts/repeatsExtractTS.sh VECTOR /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ /home/mdisk/*/00.software/path_to_location/gene-is/scripts 0.9 /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/blat /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa.2bit 95 10
awk: fatal: cannot open file `/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtered.bst' for reading (No such file or directory)
cut: /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtered.bst: No such file or directory
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/blat: error while loading shared libraries: libpng12.so.0: cannot open shared object file: No such file or directory
awk: fatal: cannot open file `/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtered.temp.bst' for reading (No such file or directory)
###################################################################
IS annotation TS...
perl -I /home/mdisk/*/00.software/path_to_location/gene-is/lib /home/mdisk/*/00.software/path_to_location/gene-is/scripts/annotation.pl -o /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -s /home/mdisk/*/00.software/path_to_location/gene-is/scripts -t /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bedtools -a1 /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/UCSC.anno.table_hg38.txt -r1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//resultsNoDup.csv.total.results
perl -I /home/mdisk/*/00.software/path_to_location/gene-is/lib /home/mdisk/*/00.software/path_to_location/gene-is/scripts/annotation.pl -o /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -s /home/mdisk/*/00.software/path_to_location/gene-is/scripts -t /home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/bedtools -a1 /home/mdisk/*/00.software/path_to_location/gene-is/test/datasets/UCSC.anno.table_hg38.txt -r1 /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//repeats.resultsNoDup.csv.total.results
Error: The requested bed file (/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ISFileMod1.bed) could not be opened. Exiting!
/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/
Error: The requested bed file (/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//anno9.bed) could not be opened. Exiting!
Error: The requested bed file (/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//anno9.bed) could not be opened. Exiting!
Error: The requested bed file (/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//ISFileMod1.bed) could not be opened. Exiting!
/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/
Error: The requested bed file (/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//anno9.bed) could not be opened. Exiting!
Error: The requested bed file (/home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//anno9.bed) could not be opened. Exiting!
###################################################################
###################################################################
Generating General Statistics ...
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/samtools: error while loading shared libraries: libncurses.so.5: cannot open shared object file: No such file or directory
Finished.
###################################################################
##################### Testing Single-end Output #########################
Generated Output File /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/testDataTS.single.csv
Template Output File /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/testDataTS.ResultsClusteredAnnotated.csv
!!! Assertion failed !!!
Output File /home/mdisk/*/00.software/path_to_location/gene-is/test/targetedSequencing/results/testDataTS.single.csv is not the same as expected
###################################################################
可以看到,该脚本也是分步处理的数据,下面我们将脚本的每一大步骤进行拆分,以熟悉针对单端测序数据的转座子插入序列分析的全流程。
第一步,进行数据预处理
perl /home/mdisk/****/00.software/path_to_location/gene-is/scripts/filteringTrimming.pl -f /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/testData.TS.pair1.fastq.gz -qual 20 -adaptF GATCGGAAGAGCACACGTCTGAACTCCAGTCAC -sOut filtTrim -o /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -sk /home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/skewer
调用了filteringTrimming.pl的功能,基本功能如下
Please provide a forward file!
Usage: filterTrimming.pl <-f forward file> <required> <-skewer full path is required> <required>. [options]
Options:
-h, --help Displays this infrOutmation.
-f, --forward Forward FASTQ file <required>.
-sk, --skewer Full path of skewer tool is needed <required>.
-r, --reverse Reverse FASTQ file.
-qual, --quality Quality value for filteration <0-40>.Default is 20
-adaptF, --adapterForward Adapter for forward file.Default is GATCGGAAGAGCACACGTCTGAACTCCAGTCAC
-adaptR, --adapterReverse Adapter for reverse file.Default is AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT
-sOut, --suffOut Name for suffix output file. Default is filtTrim
-o, --output Full path of a directory to store results.Default is current working directory.
This program quality filter and trim adapters of the provided FASTQ files
可见,该程序的主要功能是修剪指定接头的序列以及测序数据的质控。
有几个必须项和默认项,
必须项:
-f提供的5’端测序文件
-sk skewer工具的决定引用地址
默认项:
-qual, 用于序列总体质量过滤的阈值,默认是20,可选范围是0-40.
-adaptF,5’测序文件的接头,默认是GATCGGAAGAGCACACGTCTGAACTCCAGTCAC。
-sOut 输出文件的前缀,默认是filtTrim。
-o 输出文件的地址,默认是当前工作路径。
代码中可以看到,同时调用了skewer程序,我们查看一下skewer的功能,定位到上述程序的位置后输入./skewer --h即可查看,程序介绍如下:
Skewer (A fast and accurate adapter trimmer for paired-end reads)一个快速且准确的双端数据接头修剪器
Version 0.1.117 (updated in July 12, 2014), Author: Hongshan Jiang
USAGE: skewer [options] <reads.fastq> [paired-reads.fastq]
or skewer [options] - (for input from STDIN)
OPTIONS (ranges in brackets, defaults in parentheses):
Adapter:
-x <str> Adapter sequence/file (AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC)指定
-y <str> Adapter sequence/file for pair-end reads (AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTA),
implied by -x if -x is the only one specified explicitly.双端读取的适配器序列/文件(AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTA),如果-x是唯一显式指定的适配器序列/文件,则由-x指定。
-j <str> Junction adapter sequence/file for Nextera Mater Pair reads (CTGTCTCTTATACACATCTAGATGTGTATAAGAGACAG)
-m, --mode <str> trimming mode; 1) single-end -- head: 5' end; tail: 3' end; any: anywhere (tail)
2) paired-end -- pe: paired-end; mp: mate-pair (pe)指定修剪模式
Tolerance:
-r <num> Maximum allowed error rate (normalized #errors / length of aligned region) [0, 0.5], (0.1)最大允许的错误率
-d <num> Maximum allowed indel error rate [0, r], (0.03)最大允许的插入缺失的错误率
reciprocal is used for -r and -d when num > or = 2
-k <int> Minimum overlap length for adapter detection [1, inf);
(max(1, int(4-10*r)) for single-end; (<junction length>/2) for mate-pair)
Filtering & Post-trimming:
-q, --end-quality <int> Trim 3' end until specified or higher quality reached; (0)
-Q, --mean-quality <int> The lowest mean quality value allowed before trimming; (0)
-l, --min <int> The minimum read length allowed after trimming; (18)
-L, --max <int> The maximum read length allowed after trimming; (no limit)
-n Whether to filter out highly degerative (many Ns) reads; (no)
-u Whether to filter out undetermined mate-pair reads; (no)
Input/Output:
-f, --format <str> Format of FASTQ quality value: sanger|solexa|auto; (auto)
-b, --barcode Use adapters to demultiplex reads to trimmed file(s) and an untrimmed file (no)
-o, --output <str> Base name of output file; ('<reads>.trimmed-Q<int>L<int>')
-z, --compress Compress output in GZIP format (no)压缩文件为GZIP格式
-1, --stdout Redirect output to STDOUT, suppressing -b, -o, and -z options (no)
--quiet No progress update (not quiet)
Miscellaneous:
-t, --threads <int> Number of concurrent threads [1, 16]; (1)指定线程数
EXAMPLES:
skewer -Q 9 -t 2 -x adapters.fa sample.fastq -o trimmed
skewer -x AGATCGGAAGAGC -q 3 sample-pair1.fq.gz sample-pair2.fq.gz
skewer -x TCGTATGCCGTCTTCTGCTTGT -l 16 -L 30 -d 0 srna.fastq
skewer -m mp lmp-pair1.fastq lmp-pair2.fastq
我们可以看到,
#system "(fastq_quality_fiilter -q $qual_value -p $perc_value -i $f_file -o $output_dir/filt11.fastq -Q33)";
system "(echo \"$skewer_dir -x $adaptF_value -y $adaptR_value -q $qual_value -l 50 -o $output_dir/$Out_value $f_file $r_file\")";
system "($skewer_dir -x $adaptF_value -y $adaptR_value -q $qual_value -l 20 -o $output_dir/$Out_value $f_file $r_file)";
#system "(fastq_quality_filter -q $qual_value -p $perc_value -i $r_file -o $output_dir/filt22.fastq -Q33)";
}else{
system "(echo \"$skewer_dir -x $adaptF_value -q $qual_value -l 50 -o $output_dir/$Out_value $f_file\")";
system "($skewer_dir -x $adaptF_value -q $qual_value -l 20 -o $output_dir/$Out_value $f_file)";
前面给出的代码与默认基本一致,我们先看一下输入的文件的基本信息
 通过检索,可以看到很多序列都包含adapter序列,运行程序之后,可以看到生成了以下文件:
通过检索,可以看到很多序列都包含adapter序列,运行程序之后,可以看到生成了以下文件:

我们先查看一下运行日志filtTrim.log
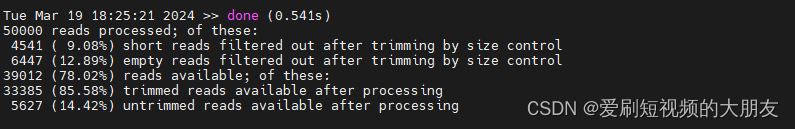
再查看一下filtTrim.fastq文件
 结合之前的接头序列的标记图,可以看到,很多序列在识别到adapter后,其下游序列均被修剪掉了,同时删除了修剪后过短的序列,继续观察可以发现,部分序列未识别到接头序列的完全匹配序列,但仍经历了修剪过程,这是因为程序对于接头序列的识别较为灵敏,因为测序过程中存在一定的错误率,较为宽容的识别可提高识别的灵敏度。
结合之前的接头序列的标记图,可以看到,很多序列在识别到adapter后,其下游序列均被修剪掉了,同时删除了修剪后过短的序列,继续观察可以发现,部分序列未识别到接头序列的完全匹配序列,但仍经历了修剪过程,这是因为程序对于接头序列的识别较为灵敏,因为测序过程中存在一定的错误率,较为宽容的识别可提高识别的灵敏度。
 因为这个脚本的基本功能是调用上面提到的那些工具(exp:samtools,bedtools等)我们还可以看一下他的脚本,具体使用了那些参数
因为这个脚本的基本功能是调用上面提到的那些工具(exp:samtools,bedtools等)我们还可以看一下他的脚本,具体使用了那些参数
system "(echo \"$skewer_dir -x $adaptF_value -q $qual_value -l 50 -o $output_dir/$Out_value $f_file\")";
system "($skewer_dir -x $adaptF_value -q $qual_value -l 20 -o $output_dir/$Out_value $f_file)"
可以看到,脚本主要使用了skewer的-x,-q,-l和-o参数
-x 是指定需要修剪的序列,如果没有指定则默认AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC序列,脚本中更换为了GATCGGAAGAGCACACGTCTGAACTCCAGTCAC 序列;
-q是对末端(3’ end)进行修剪,直到达到指定的质量阈值或获得更高质量的序列。
-l
在数据修剪(质控)之后,开始运行比对程序,使用的是BWA
###################################################################
Alignment in process...
=======>>>>>>>>>>>> 0
perl -I /home/mdisk/****/00.software/path_to_location/gene-is/lib /home/mdisk/****/00.software/path_to_location/gene-is/scripts/alignment.pl -p 8 -f filtTrim.fastq -gv /home/mdisk/****/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa -a /home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/bwa -aOut completAlignment -o /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd/ -t AGILENT -sam /home/mdisk/*****/00.software/path_to_location/gene-is/tools/bin/samtools
Alignemnt type AGILENT
BWA is used as Aligner
/home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/bwa mem -M -t 8 /home/mdisk/****/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq > /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//completAlignment.sam
[M::main_mem] read 39012 sequences (6961052 bp)...
[main] Version: 0.7.4-r385
[main] CMD: /home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/bwa mem -M -t 8 /home/mdisk/****/00.software/path_to_location/gene-is/test/datasets/testGenomeVector.fa /home/mdisk/****/00.software/path_to_location/gene-is/test/targetedSequencing/results/singleEnd//filtTrim.fastq
[main] Real time: 0.902 sec; CPU: 5.069 sec
/home/mdisk/****/00.software/path_to_location/gene-is/tools/bin/samtools: error while loading shared libraries: libncurses.so.5: cannot open shared object file: No such file or directory
/home/mdisk/*/00.software/path_to_location/gene-is/tools/bin/samtools: error while loading shared libraries: libncurses.so.5: cannot open shared object file: No such file or directory
Perl 模块
所需的 Perl 库被预先打包在工具中(GENE-IS 中的“ lib”dir)。
配置文件
GENE-IS 拥有针对每种分析模式的特定配置文件; LAM-PCR、 TES 配对和 TES 单端配置文件。只有相关的配置文件应该为特定的分析进行修改。为了测试 GENE-IS 安装,用户不需要更改配置文件中的任何参数。模板位于基因路径中,即。
$GENIS/configFile_targetedSequencing_pairedEnd.txt
Contacts
Contact: raffaele.fronza@nct-heidelberg.de
Contact: saira.afzal@genewerk.de















 334
334











 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









